很久很久以前我就写过一个服务器系列教程:http://www.bio-info-trainee.com/555.html
在那里,我留下了一个疑问,因为后来没有机会再继续捣鼓服务器,所以一直悬而未决,问题描述如下: Continue reading
三
24
很久很久以前我就写过一个服务器系列教程:http://www.bio-info-trainee.com/555.html
在那里,我留下了一个疑问,因为后来没有机会再继续捣鼓服务器,所以一直悬而未决,问题描述如下: Continue reading
一般来说看链接最后的文件名就知道这篇文章讲的是什么了:
这本是我为论坛的基础板块写的一个基础知识点,但是浏览量实在有限,不忍它蒙尘,特在博客重新发布一次!原帖见:http://www.biotrainee.com/thread-511-1-1.html
gene symbol 是非常官方的,由HUGO 组织负责维护,有专门的数据库HGNC database of human gene names | HUGO
以前分析数据的时候,有一些基因的symbol很奇怪,让我百思不得其解,比如
C orf 系列基因,
HS.系列基因,
KRTAP系列基因,
LOC系列基因,
MIR系列基因,
LINC系列基因
它们往往一个系列,就有好几百个基因;
C12orf44; Chromosome 12 Open Reading Frame 44; 这个是C orf系列基因的意思
MIR系列基因应该是 miRNA相关的基因
LINC系列基因应该就是long intergenic non-protein coding RNA
LOC系列基因,是非正式的,推定的,日后可能被更合适的名字替代
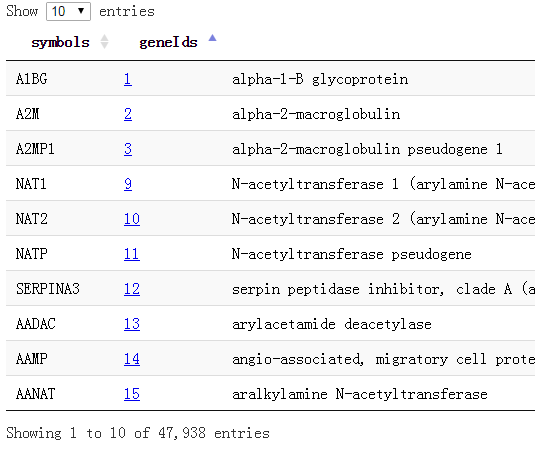
我这里做好了所有的基因对应关系,去生信菜鸟团QQ群里下载吧,共47938个基因的symbol和entrez gene id还有name,还有alias的对应!
还有一些RNA基因,根本就没有symbol,比如:CTA/B/C/D系列的
Aliases for ENSG00000271971 Gene
Quality Score for this RNA gene is 1
Aliases for ENSG00000271971 Gene
CTD-2006H14.2 5
External Ids for ENSG00000271971 Gene
Ensembl: ENSG00000271971
还有,如果你看到HS.开头的基因,它是unigene的ID了,已经不再是symbol啦。
这是系列文章,请先看:
ncbi现有的GPL已经过万了,但是bioconductor的芯片注释包不到一千,虽然bioconductor可以解决我们大部分的需要,比如affymetrix的95,133系列,深圳1.0st系列,HTA2.0系列,但是如果碰到比较生僻的芯片,bioconductor也不会刻意为之制作一个bioconductor的包,这时候就需要自行下载NCBI的GPL信息了,也可以通过R来解决:
##本质上是下载一个文件,读进R里面,然后解析行列式,得到芯片探针与基因的对应关系,看下面的代码,你就能理解了。 Continue reading
首先要明白这个ES score图片里面的数据是什么,这样才能修改它,因为java是一个封闭打包好的软件,所以我们没办法在里面修改它没有提供的参数,运行完GSEA,默认输出的图就是下面这样: Continue reading
我以为我写完了R包终极解决方案! 之后,应该不会再有任何关于R包安装的问题产生了,但仔细回过头来看才发现,我介绍的都是如何从CRAN或者bioconductor里面安装正规发布的包,但是有很多人开发的是自己私人的包,而我们有的确非常需要用怎么办??这个时候就需要下载别人开发的包来安装了。比如我R包地址见github:https://github.com/jmzeng1314/humanid Continue reading
随着R语言的流行度的提高,开发一个R包已经不再是专业程序猿才有的技能了。我这里讲的不是如何写一个包含了复杂统计公式或者发表一篇SCI文章的包,而是简简单单的用Rstudio自带的创建包的功能把自己的几个函数和数据打包!!!我R包地址见github:https://github.com/jmzeng1314/humanid Continue reading
经过热心的小伙伴的提醒,我才知道我以前写的R语言画网络图三部曲竟然漏掉了最基础的一个包,就是igraph,不了解这个,后面的两个也是无源之水。
对于生物出身的部分生物信息学工程师来说,很多计算机概念让人很头疼,尤其是计算机语言里面的高级对象。我以前学编程的时候,给我一个变量,一个数据,一个hash,我就心满意足了,可以解决大部分我数据处理问题,可事情远比想象之中复杂。因为很多高手喜欢用封装,代码复用,喜欢用高级对象。在R的bioconductor里面尤其是如此,经常会遇到各种包装好的S3,S4对象,看过说明书,倒是知道一些对象里面有什么,可以去如何处理那些对象,提取我们想要的信息,比如我就写过一系列的帖子:
因为买过一个超算云服务器,所以前面我讲过Ubuntu服务器管理系列知识,正好最近要搞了个阿里云,用来做shiny服务器,发现服务器管理居然进化了好多,以前的知识都过时了,再记录一笔吧,真的是学习如逆水行舟,不进则退呀!
我的阿里云服务器版本是CentOS 6.5.,属于(RedHat 7, Ubuntu 15.04+, SLES 12+) 系列,是目前最新版本的服务器管理,所以大家重点是记住这个systemctl 即可:
如果你已经安装好了shiny 服务器,(安装教程)要开始使用了,掌握一些基础知识是必须的。这里我简单学习了一些入门资料,分享给大家,慢慢的我会写一个进阶资料。安装成功之后,系统会增加4个目录,是一定要掌握的:
1、这个目录只存放关键配置文件:/etc/shiny-server/shiny-server.conf 初始状态只有一个文件,记录着非常多的默认信息,默认的网站目录是根目录下的srv的shiny-server目录,端口是3838
2、网站运行log日子存放:/var/log/shiny-server 初始状态下该目录为空3、程序存放目录是:/srv/shiny-server 初始状态,有一个测试程序:
4、最后是/opt/shiny-server/ 目录,这里面也有一个配置文件:/opt/shiny-server/config/default.config
个人比较欣赏R shiny制作的网页,入门简单,上手极快,多看点例子,制作复杂逻辑的网页也不是问题。这篇实战指南有四个步骤:
至少需要root权限的linux系统 (我测试了阿里云)
安装R (一般安装最新版,)
在R中安装shiny模块 (一般还可以多安装一些模块)
下载并且安装shiny server安装包 (根据系统选择)
R studio公司毕竟是商业化公司,在R语言推广方面做得很棒。网站什么总共有9个cheatsheet,R语言入门完全可以把这个当做笔记,写代码随时查用!
我批量下载了所有,但是想打印的时候,发现挺麻烦的,因为我不知道批量打印的方法,索性我还是半个程序猿,所以搜索了一下批量合并pdf的方法,这样就可以批量打印了,也方便传输这个文件。
其实如果在linux系统里面,一般都会自带pdf toolkit工具,里面有命令可以合并PDF文档。 Continue reading
我前面写到了生信分析人员如何入门linux和perl,后面还会写R和python的总结,但是在这中间我想插入一个脚本实战指南。其实在我前两篇日志里面也重点提到了学习编程语言最重要的就是实战了,也点出了几个关键词。在实际生物信息学数据处理中应用perl和linux,可以借鉴EMBOSS软件套件,fastx-toolkit等基础软件,实现并且模仿该软件的功能。尤其是SMS2/exonerate/里面的一些常见功能,还有DNA2.0 Bioinformatics Toolbox的一些工具。如果你这些名词不懂,请赶快谷歌!!! 它们做了什么,输入文件是什么,输出文件是什么,你都可以用脚本实现!
R与ASReml-R统计分析教程(林元震)中国林业出版社
1-3章简单介绍了R的基本语法,然后第4章着重讲了各种统计方法,第5章讲R的绘图,最后一张讲ASReml-R这个包
语法重点:
1,install.packages(),library(),help(),example(),demo(),length(),attribute(),class(),mode(),dim(),names(),str(),head(),
tail()
2,rep,seq,paste,array,matrix,data.frame,list,c(),factor(),
3,缺失值处理(na.omit,na.rm=T),类型转换(as.numeric(),as.character(),as.factor(),as.logical())
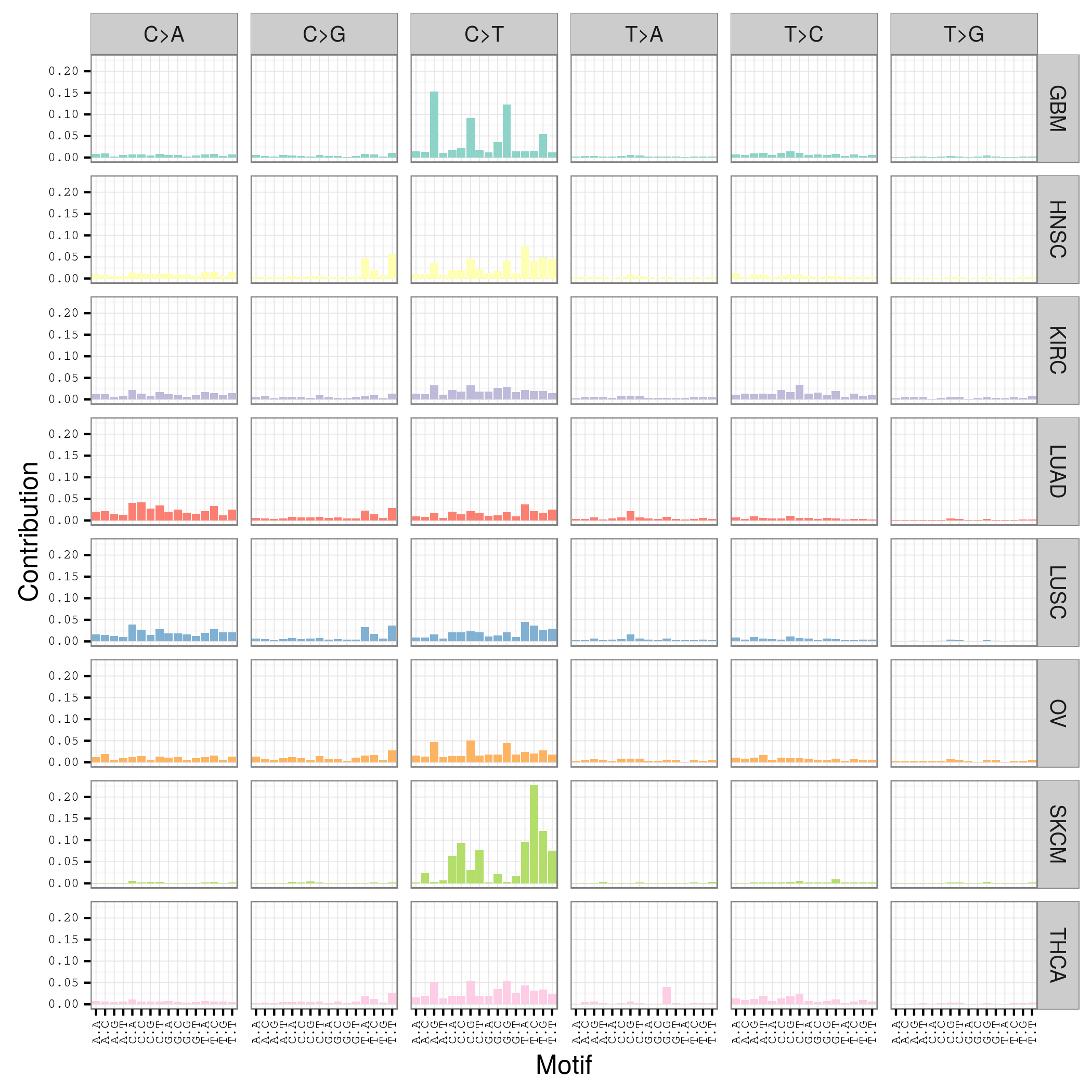
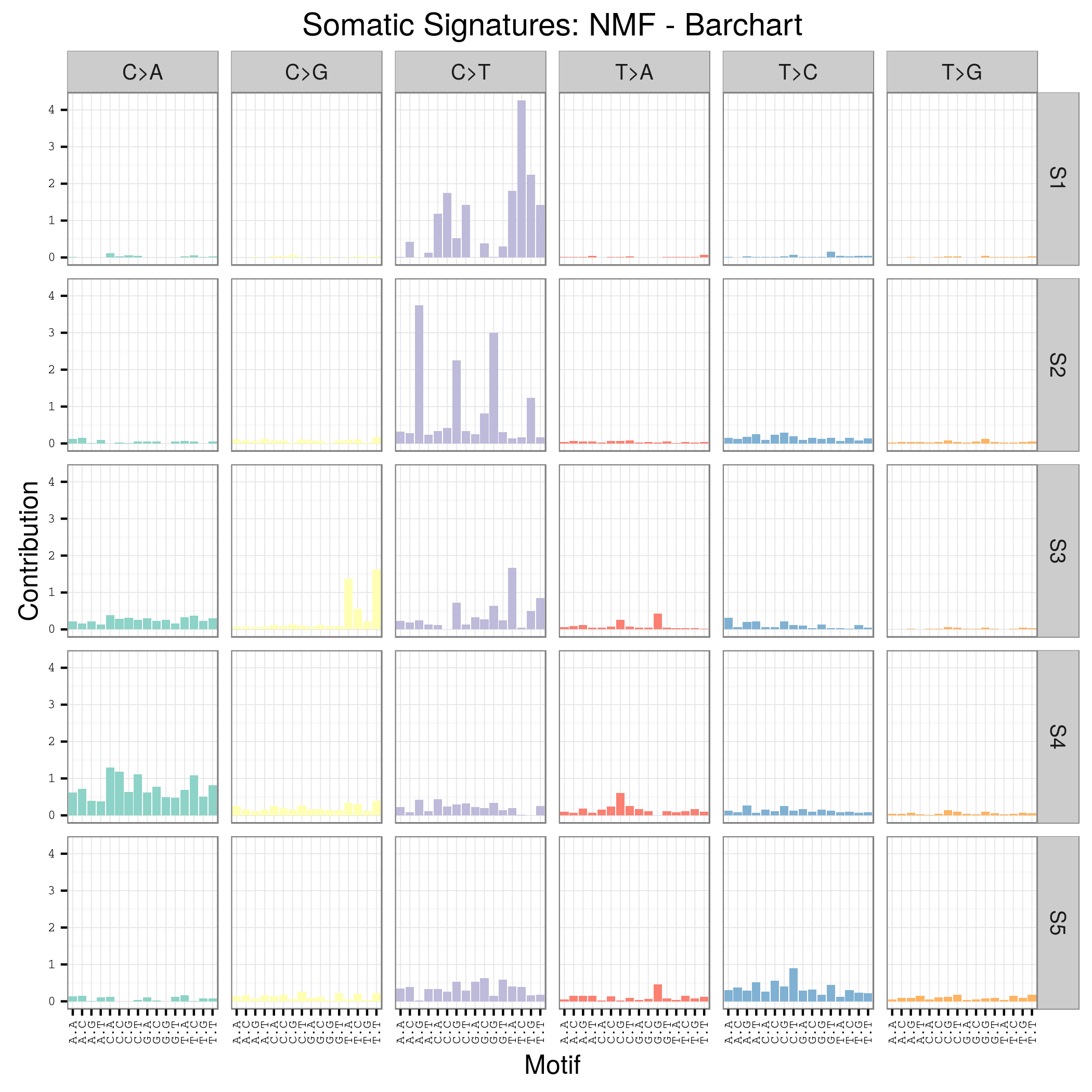
mutation signature这个概念提出来还不久,我看了看文献,最早见于2013年的一篇nature文章,主要是用来描述癌症患者的somatic mutation情况的。
首先要自己分析癌症样本数据,拿到somatic mutation,TCGA计划发展到现在已经有非常多的somatic mutation结果啦,大家可以自行选择感兴趣的癌症数据拿来研究,解析一下mutation signature 。
我这里给大家推荐一个工具,是R语言的Bioconductor系列包中的一个,SomaticSignatures
其实它的说明书写的非常详细了已经,如果你理解了mutation signature的概念,很容易用那个包,其实你自己写一个脚本也是非常任意的,就是根据mutation的位置在基因组中找到它的前后一个碱基,然后组成三碱基突变模式,最后统计一下那96种突变模式的分布状况!
我这里简单讲一讲这个包如何用吧!
首先下载并加载几个必须的包:
然后根据突变数据做好一个GRanges对象,这个可以看我以前的博客