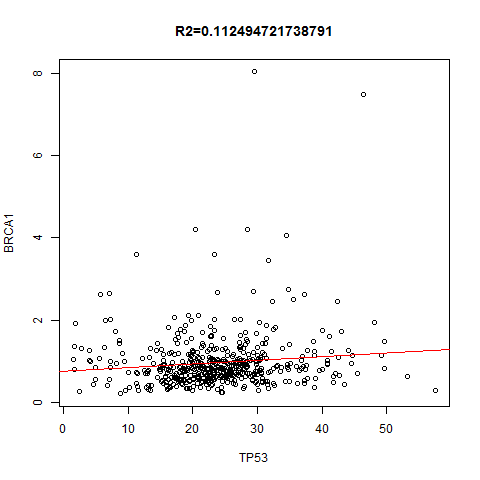
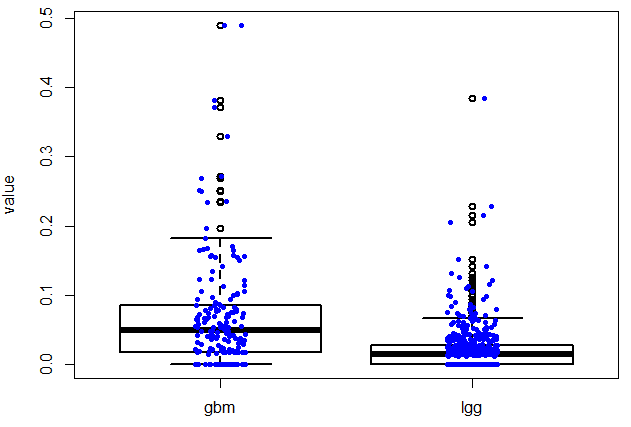
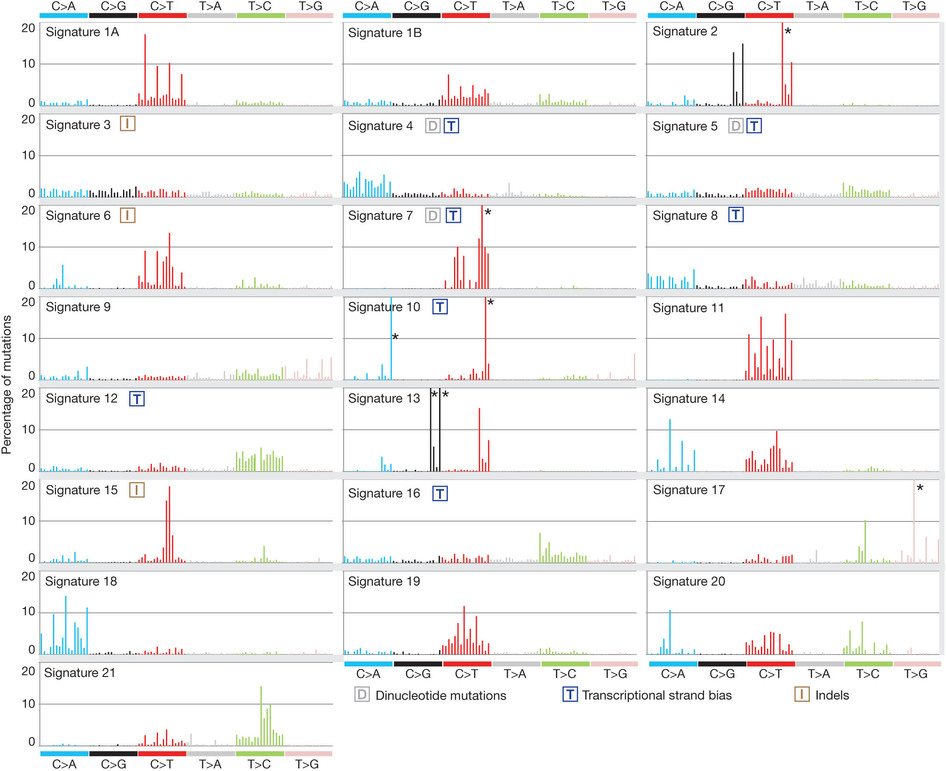
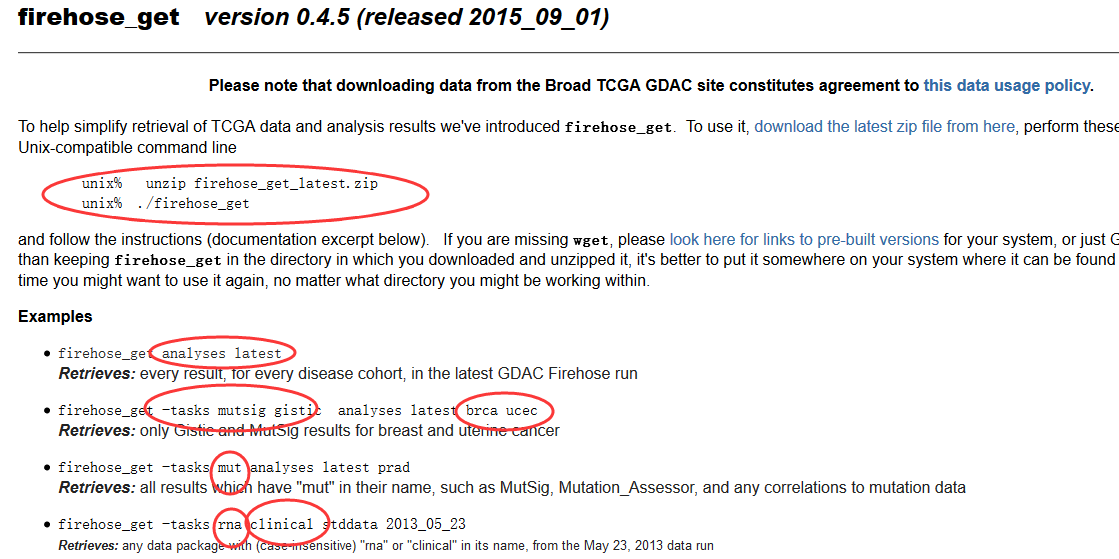
TCGA想必搞生信都或有耳闻,尤其是癌症研究方向的,共4个年度研讨会,主要是pdf格式的ppt分享,有需要的可以具体点击到页面一个个下载自己慢慢研究,也可以用我下面链接直接下载。
本来是有youtube分享演讲视频的,但是国内被墙了,大家就看看ppt吧
http://www.genome.gov/17516564
The Cancer Genome Atlas (TCGA) is a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer through the application of genome analysis technologies, including large-scale genome sequencing.
TCGA is a joint effort of the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI), which are both part of the National Institutes of Health, U.S. Department of Health and Human Services.
Meetings
pdf链接地址如下
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Laird.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Durbin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Ley.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Sartor.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Ciriello.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Imielinski.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Gao.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Carter.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Ng.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Parvin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Raphael.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Lawrence.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Kreisberg.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Marra.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Helman.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Stuart.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Cooper.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Levine.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Natsoulis.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Haussler.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Erkkila.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Gehlenborg.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Qiao.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Sivachenko.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Sumazin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Gutman.pdf
http://www.genome.gov/Multimedia/Slides/TCGA1/TCGA1_Mardis.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/01_Shaw.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/02_Chanock.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/03_Staudt.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/05_Creighton.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/06_Stojanov.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/07_Karchin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/08_Mungall.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/09_Hakimi.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/10_Gao.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/11_Hayes.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/12_Troester.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/13_Knobluach.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/14_Raphael.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/15_Akbani.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/16_Giordano.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/17_Weinstein.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/18_Zheng.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/19_Getz.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/20_VanDneBroek.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/21_Liao.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/22_Khazanov.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/23_Levine.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/24_Miller.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/25_Ewing.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/26_Cirello.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/27_Verhaak.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/28_Hofree.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/29_Meyerson.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/30_Yang.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/31_Wheeler.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/32_Parfenov.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/33_Bernard-Rovira.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/34_Hast.pdf
http://www.genome.gov/Multimedia/Slides/TCGA2/36_Sellars.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/04_Brat.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/05_Mungall.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/06_Boutros.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/07_Zmuda.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/08_Benz.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/09_Zheng.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/11_Creighton.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/12_Aksoy.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/13_Dinh.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/14_Stuart.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/15_Amin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/16_Gross.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/15_Akbani.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/18_Giordano.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/19_Amin-Mansour.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/20_Oesper.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/21_Gatza.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/22_Bernard.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/23_Sinha.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/24_Akbani.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/25_Watson.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/26_Martignetti.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/27_Bandlamudi.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/28_Fu.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/29_Akdemir.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/30_Bass.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/31_Hakimi.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/32_Wheeler.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/33_Lehmann.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/34_Gordenin.pdf
http://www.genome.gov/Multimedia/Slides/TCGA3/35_Wyczalkowski.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/02_Zenklusen.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/03_Hutter.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/04_Brat.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/05_Mungall.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/06_Linehan.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/07_Brooks.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/08_Wu.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/09_Giger.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/10_Wilkerson.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/11_Orsulic.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/12_Zhong.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/13_Knijnenburg.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/14_Akbani.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/15_Wang.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/16_Poisson.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/17_Alaeimahabadi.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/18_Noushmehr.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/19_Pantazi.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/20_Shih.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/21_Stransky.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/22_Giordano.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/23_Davidsen.pdf
http://www.genome.gov/Multimedia/Slides/TCGA4/24_Gross.pdf