最近在忙一些chip-seq的数据分析项目,它的可视化展现比较复杂一点,自己写程序将会耗费挺长时间的,就想着利用现成的工具,前面试用了deeptools,挺好 的,但是有点慢,是python程序,如下:
现在换一个R程序,这个非常快速,而且绘图个人觉得稍微美观一点,大家也可以都试试看。
首先软件的github里面有源代码,然后作者还四处宣讲这个包的神奇之处,下面的ppt非常言简意赅的描述了它的功能和强大之处。
对于CHIP-seq数据处理完全是自学的,所以有很多细节得慢慢学习回来,这次记录的就是当我们把测序仪的fastq数据比对到参考基因组之后,应该对比对的结果文件做什么样的处理,然后去给peaks caller软件拿来call peaks呢?我看过博客 提到只保留比对质量值大于30的,也看过博客提到只保留unique比对的reads,我这里拿一篇公共数据测试了一下它们的区别!数据描述如下: Continue reading
之前分析了好几个公共项目,拿到的peaks都很诡异,搞得我一直怀疑是不是自己分析错了。终于,功夫不负有心人,我分析了一个数据,它的peaks非常完美!!!可以证明,我的分析流程以及peaks绘图代码并没有错!数据来自于http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74311,是关于H3K27ac_ChIP-Seq_LOUCY,组蛋白修饰的CHIP-seq数据,很容易就下载了作者上传的测序数据,然后跑了我的流程!https://github.com/jmzeng1314/NGS-pipeline/tree/master/CHIPseq Continue reading
讲到这里,我们的自学CHIP-seq分析系列教程就告一段落了,当然,我会随时查漏补缺,根据读者的反馈来更新着系列教程。其实可视化这已经是一个比较复杂的方向了,不仅仅是针对于CHIP-seq数据。可视化本身是发文章的先决条件,而让人一目了然图片也说明了数据分析人员对数据本身的理解。我这里就列出一些目录和一些工具,和ppt。这个主要靠大家自学了,而且我博客空间有限,就不上传一大堆图片了,大家随便找一些经典的paper里面都会有很多可视化分析。
首先强烈推荐两个网页版工具,针对找到的peaks可视化:
然后再推荐一个哈佛刘小乐实验室出品的软件,也是专门为了作图http://liulab.dfci.harvard.edu/CEAS/usermanual.html
经过前面的CHIP-seq测序数据处理的常规分析,我们已经成功的把测序仪下机数据变成了BED格式的peaks记录文件,我选取的这篇文章里面做了4次CHIP-seq实验,分别是两个重复的野生型MCF7细胞系的 BAF155 immunoprecipitates和两个重复的突变型MCF7细胞系的 BAF155 immunoprecipitates,这样通过比较野生型和突变型MCF7细胞系的 BAF155 immunoprecipitates的结果的不同就知道该细胞系的BAF155 突变,对它在全基因组的结合功能的影响啦。
#我这里直接从GEO里面下载了peaks结果,它们详情如下:wc -l *bed
6768 GSM1278641_Xu_MUT_rep1_BAF155_MUT.peaks.bed
3660 GSM1278643_Xu_MUT_rep2_BAF155_MUT.peaks.bed
11022 GSM1278645_Xu_WT_rep1_BAF155.peaks.bed
5260 GSM1278647_Xu_WT_rep2_BAF155.peaks.bed
49458 GSM601398_Ini1HeLa-peaks.bed
24477 GSM601398_Ini1HeLa-peaks-stringent.bed
12725 GSM601399_Brg1HeLa-peaks.bed
12316 GSM601399_Brg1HeLa-peaks-stringent.bed
46412 GSM601400_BAF155HeLa-peaks.bed
37920 GSM601400_BAF155HeLa-peaks-stringent.bed
30136 GSM601401_BAF170HeLa-peaks.bed
25432 GSM601401_BAF170HeLa-peaks-stringent.bed
每个BED的peaks记录,本质是就3列是需要我们注意的,就是染色体,以及在该染色体上面的起始和终止坐标,如下:
#PeakID chr start end strand Normalized Tag Count region size findPeaks Score Clonal Fold Change
chr20 52221388 52856380 chr20-8088 41141 +
chr20 45796362 46384917 chr20-5152 31612 +
chr17 59287502 59741943 chr17-2332 29994 +
chr17 59755459 59989069 chr17-667 19943 +
chr20 52993293 53369574 chr20-7059 12642 +
chr1 121482722 121485861 chr1-995 9070 +
chr20 55675229 55855175 chr20-6524 7592 +
chr3 64531319 64762040 chr3-4022 7213 +
chr20 49286444 49384563 chr20-4482 6165 +
我们所谓的peaks注释,就是想看看该peaks在基因组的哪一个区段,看看它们在各种基因组区域(基因上下游,5,3端UTR,启动子,内含子,外显子,基因间区域,microRNA区域)分布情况,但是一般的peaks都有近万个,所以需要批量注释,如果脚本学的好,自己下载参考基因组的GFF注释文件,完全可以自己写一个,我这里会介绍一个R的bioconductor包ChIPpeakAnno来做CHIP-seq的peaks注释,下面的包自带的示例:
library(ChIPpeakAnno)
bed <- system.file("extdata", "MACS_output.bed", package="ChIPpeakAnno")
gr1 <- toGRanges(bed, format="BED", header=FALSE)
## one can also try import from rtracklayer
library(rtracklayer)
gr1.import <- import(bed, format="BED")
identical(start(gr1), start(gr1.import))
gr1[1:2]
gr1.import[1:2] #note the name slot is different from gr1
gff <- system.file("extdata", "GFF_peaks.gff", package="ChIPpeakAnno")
gr2 <- toGRanges(gff, format="GFF", header=FALSE, skip=3)
ol <- findOverlapsOfPeaks(gr1, gr2)
makeVennDiagram(ol)##还可以用binOverFeature来根据特定的GRanges对象(通常是TSS)来画分布图
## Distribution of aggregated peak scores or peak numbers around transcript start sites.
可以看到这个包使用起来非常简单,只需要把我们做好的peaks文件(GSM1278641_Xu_MUT_rep1_BAF155_MUT.peaks.bed等等)用toGRanges或者import读进去,成一个GRanges对象即可,上面的代码是比较两个peaks文件的overlap。然后还可以根据R很多包都自带的数据来注释基因组特征:
data(TSS.human.GRCh37) ## 主要是借助于这个GRanges对象来做注释,也可以用getAnnotation来获取其它GRanges对象来做注释
## featureType : TSS, miRNA, Exon, 5'UTR, 3'UTR, transcript or Exon plus UTR
peaks=MUT_rep1_peaks
macs.anno <- annotatePeakInBatch(peaks, AnnotationData=TSS.human.GRCh37,
output="overlapping", maxgap=5000L)## 得到的macs.anno对象就是已经注释好了的,每个peaks是否在基因上,或者距离基因多远,都是写的清清楚楚
if(require(TxDb.Hsapiens.UCSC.hg19.knownGene)){
aCR<-assignChromosomeRegion(peaks, nucleotideLevel=FALSE,
precedence=c("Promoters", "immediateDownstream",
"fiveUTRs", "threeUTRs",
"Exons", "Introns"),
TxDb=TxDb.Hsapiens.UCSC.hg19.knownGene)
barplot(aCR$percentage)
}
得到的条形图如下,虽然很丑,但这就是peaks注释的精髓,搞清楚每个peaks在基因组的位置特征:
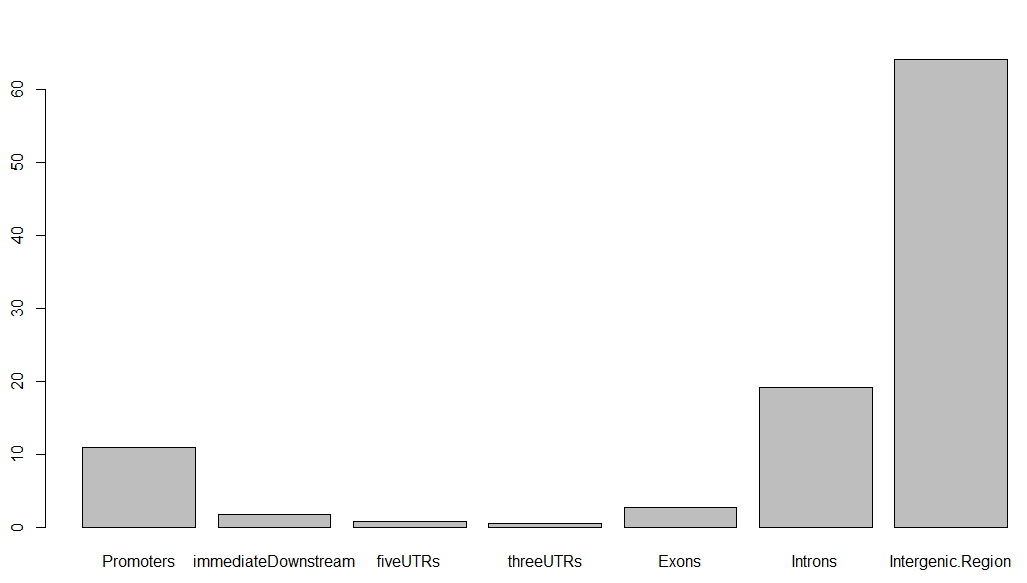
同理,对每个peaks文件,都可以做类似的分析!
但是对多个peaks文件,比如本文中的,想比较野生型和突变型MCF7细胞系的 BAF155 immunoprecipitates的结果的不同,就需要做peaks之间的差异分析,已经后续的差异基因注释啦
当然,值得一提的是peaks注释我更喜欢网页版工具,反正peaks文件非常小,直接上传到别人做好的web tools,就可立即出一大堆可视化图表分析结果啦,大家可以去试试看:
BayesPeak也是peaks caller家族一员,用的人也不少,我这次也试了一下,因为是R的bioconductor系列包,所以直接在R里面安装就好,但是有几个点需要注意,我比对的基因组不只是Chr1~22,X,Y,M,还有一些contig和scaffold,需要在bam文件里面去除的,而且BayesPeak比较支持读取BED文件,可以直接转为GRanges对象,虽然它号称可以使用多核,但是计算速度还是非常慢。 Continue reading
此文专门讲这个软件如何用,但是跟我以前写的软件说明书又不大一样,主要是因为我用MACS2这个软件call peaks并没有达到预期的结果,所以就多使用了几个软件,其中PeakRanger尤其值得一提,安装特别简单,而且处理数据的速度特别快,结果也非常容易理解,更重要的是它给出一个网页版的报告,里面有所有找到的符合要求的peaks的可视化图片!!!! Continue reading
CHIP-seq测序的本质还是目标片段捕获测序,跟WES不同的是,它不是通过固定的芯片探针来固定的捕获基因组上面特定序列,而是根据你选择的IP不同,你细胞或者机体状态不同,捕获到的序列差异很大!而我们研究的重点,就是捕获到的差异。而我们对CHIP-seq测序数据寻找peaks的本质就是得到所有测序数据比对在全基因组之后在正个基因组上面的测序深度里面寻找比较突出的。比如对WES数据来说,各个外显子,或者外显子的5端到3端,理论上测序深度应该是一致的,都是50X~200X,画一个测序深度曲线,应该是近似于一条直线。对我们的CHIP-seq测序数据来说,在所捕获的区域上面,理论上测序深度是绝对不一样的,应该是近似于一个山峰。而那些覆盖度高的地方,山顶,就是我们的IP所结合的热点,也就是我们想要找的peaks,在IGV里面看到大致是下面这样:
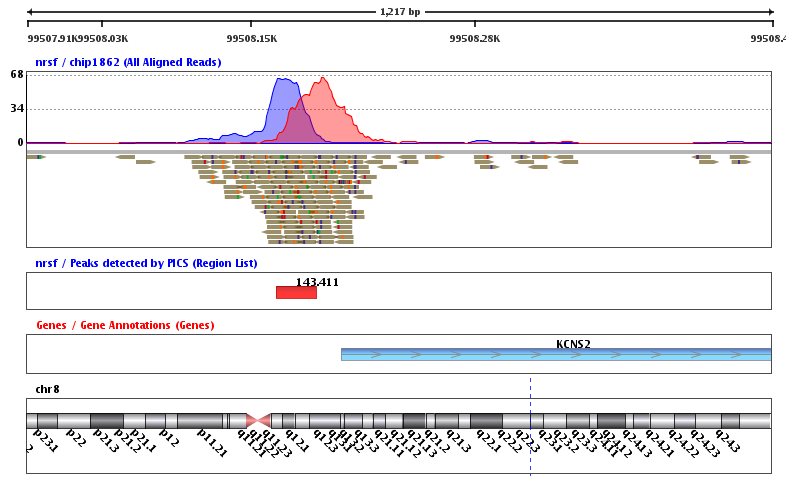
可以看到测序的reads分布是绝对的不均匀的!我们通常说的CHIP-seq测序的IP,可以是各个组蛋白的各个修饰位点对应的抗体,或者是各种转录因子的抗体,等等
如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的read所覆盖(我们测的是一个细胞群体,read出现次数多,说明该位置被TF结合的几率大)。那么,read数达到多少才叫多?这就要用到统计检验喽。假设TF在基因组上的分布是没有任何规律的,那么,测序得到的read在基因组上的分布也必然是随机的,某个碱基上覆盖的read的数目应该服从二项分布。
具体统计学原理直接看原创吧:http://www.plob.org/2014/05/08/7227.html
比对本质是是很简单的了,各种mapping工具层出不穷,我们一般常用的就是BWA和bowtie了,我这里就挑选bowtie2吧,反正别人已经做好了各种工具效果差异的比较,我们直接用就好了,代码如下:
## step5 : alignment to hg19/ using bowtie2 to do alignment
## ~/biosoft/bowtie/bowtie2-2.2.9/bowtie2-build ~/biosoft/bowtie/hg19_index /hg19.fa ~/biosoft/bowtie/hg19_index/hg19
## cat >run_bowtie2.sh
ls *.fastq | while read id ;
do
echo $id
#~/biosoft/bowtie/bowtie2-2.2.9/bowtie2 -p 8 -x ~/biosoft/bowtie/hg19_index/hg19 -U $id -S ${id%%.*}.sam 2>${id%%.*}.align.log;
#samtools view -bhS -q 30 ${id%%.*}.sam > ${id%%.*}.bam ## -F 1548 https://broadinstitute.github.io/picard/explain-flags.html
# -F 0x4 remove the reads that didn't match
samtools sort ${id%%.*}.bam ${id%%.*}.sort ## prefix for the output
# samtools view -bhS a.sam | samtools sort -o - ./ > a.bam
samtools index ${id%%.*}.sorted.bam
done
这个索引~/biosoft/bowtie/hg19_index/hg19需要自己提取建立好,见前文
初步比对的sam文件到底该如何过滤,我查了很多文章都没有给出个子丑寅卯,各执一词,我也没办法给大家一个标准,反正我测试了好几种,看起来call peaks的差异不大,就是得不到文章给出的那些结果!!
一般来说,初步比对的sam文件只能选取unique mapping的结果,所以我用了#samtools view -bhS -q 30,但是结果并没什么改变,有人说是peak caller这些工具本身就会做这件事,所以取决于你下游分析所选择的工具。
给大家看比对的日志吧:
SRR1042593.fastq
16902907 reads; of these:
16902907 (100.00%) were unpaired; of these:
667998 (3.95%) aligned 0 times
12467095 (73.76%) aligned exactly 1 time
3767814 (22.29%) aligned >1 times
96.05% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042594.fastq
60609833 reads; of these:
60609833 (100.00%) were unpaired; of these:
9165487 (15.12%) aligned 0 times
39360173 (64.94%) aligned exactly 1 time
12084173 (19.94%) aligned >1 times
84.88% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042595.fastq
14603295 reads; of these:
14603295 (100.00%) were unpaired; of these:
918028 (6.29%) aligned 0 times
10403045 (71.24%) aligned exactly 1 time
3282222 (22.48%) aligned >1 times
93.71% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042596.fastq
65911151 reads; of these:
65911151 (100.00%) were unpaired; of these:
10561790 (16.02%) aligned 0 times
42271498 (64.13%) aligned exactly 1 time
13077863 (19.84%) aligned >1 times
83.98% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042597.fastq
22210858 reads; of these:
22210858 (100.00%) were unpaired; of these:
1779568 (8.01%) aligned 0 times
15815218 (71.20%) aligned exactly 1 time
4616072 (20.78%) aligned >1 times
91.99% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042598.fastq
58068816 reads; of these:
58068816 (100.00%) were unpaired; of these:
8433671 (14.52%) aligned 0 times
37527468 (64.63%) aligned exactly 1 time
12107677 (20.85%) aligned >1 times
85.48% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042599.fastq
24019489 reads; of these:
24019489 (100.00%) were unpaired; of these:
1411095 (5.87%) aligned 0 times
17528479 (72.98%) aligned exactly 1 time
5079915 (21.15%) aligned >1 times
94.13% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042600.fastq
76361026 reads; of these:
76361026 (100.00%) were unpaired; of these:
8442054 (11.06%) aligned 0 times
50918615 (66.68%) aligned exactly 1 time
17000357 (22.26%) aligned >1 times
88.94% overall alignment rate
[samopen] SAM header is present: 93 sequences.
可以看到比对非常成功!!!我这里就不用表格的形式来展现了,毕竟我又不是给客户写报告,大家就将就着看吧。
博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
## step1 : download raw datacd ~mkdir CHIPseq_test && cd CHIPseq_testmkdir rawData && cd rawData## batch download the raw data by shell script :for ((i=593;i<601;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP033/SRP033492/SRR1042$i/SRR1042$i.sra;done
621M Jun 27 14:03 SRR1042593.sra (16.9M reads)2.2G Jun 27 15:58 SRR1042594.sra (60.6M reads)541M Jun 27 16:26 SRR1042595.sra (14.6M reads)2.4G Jun 27 18:24 SRR1042596.sra (65.9M reads)814M Jun 27 18:59 SRR1042597.sra (22.2M reads)2.1G Jun 27 20:30 SRR1042598.sra (58.1M reads)883M Jun 27 21:08 SRR1042599.sra (24.0M reads)2.8G Jun 28 11:53 SRR1042600.sra (76.4M reads)
## step2 : change sra data to fastq files.## cell line: MCF7 // Illumina HiSeq 2000 // 50bp // Single ends // phred+33ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump $id;donerm *sra
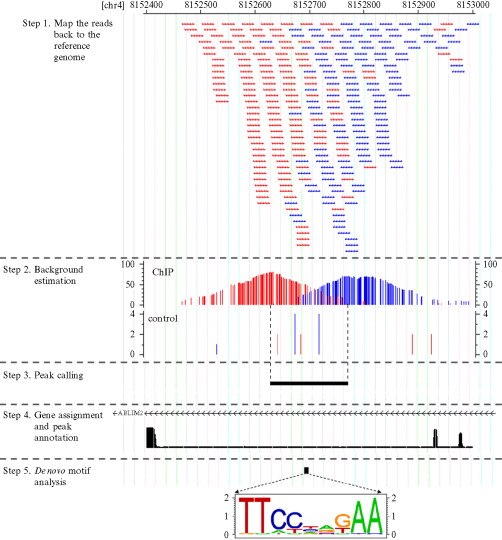
最近学习CHIP-seq的分析流程,略有点心得,也跟以前掌握的WES和RNA-seq做了一些比较,趁跑步的时候跟师妹讨论了一下,正好师妹写了一篇博客来分享这个讨论结果,我也借此机会转载过来,分享给大家,算是借花献佛吧!师妹的博文是用markdown写作,我觉得大家应该直接看她的文章,写得条理清楚:Exome-seq、ChIP-seq、RNA-seq之间的差异 Continue reading