因为买过一个超算云服务器,所以前面我讲过Ubuntu服务器管理系列知识,正好最近要搞了个阿里云,用来做shiny服务器,发现服务器管理居然进化了好多,以前的知识都过时了,再记录一笔吧,真的是学习如逆水行舟,不进则退呀!
我的阿里云服务器版本是CentOS 6.5.,属于(RedHat 7, Ubuntu 15.04+, SLES 12+) 系列,是目前最新版本的服务器管理,所以大家重点是记住这个systemctl 即可:
因为买过一个超算云服务器,所以前面我讲过Ubuntu服务器管理系列知识,正好最近要搞了个阿里云,用来做shiny服务器,发现服务器管理居然进化了好多,以前的知识都过时了,再记录一笔吧,真的是学习如逆水行舟,不进则退呀!
我的阿里云服务器版本是CentOS 6.5.,属于(RedHat 7, Ubuntu 15.04+, SLES 12+) 系列,是目前最新版本的服务器管理,所以大家重点是记住这个systemctl 即可:
pwd/ls/cd/mv/rm/cp/mkdir/rmdir/man/locate/head/tail/less/morecut/paste/join/sort/uniq/wc/cat/diff/cmp/aliaswget/ssh/scp/curl/ftp/lftp/mysql/
软硬链接区别文本编辑,文件权限设置打包压缩解压操作(tar/gzip/bzip/ x-j x-c vf)软件的快捷方式如何实现?软件如何安装(源码软件,二进制可执行软件,perl/R/python/java软件)软件版本如何管理,各种编程语言环境如何管理,模块如何管理?(尤其是大部分没有root权限)
二是shell脚本,类似于windows的bat批处理文件
三是高级运维技巧
matlab毕竟是收费软件,而且是有界面的。所以搞生物信息的都用R和linux替代了,但是很多高大上的单位,比如大名鼎鼎的broadinstitute,是用matlab的,所以他们开发的程序也会以matlab代码的形式发布。但是考虑到大多研究者用不起matlab,或者不会用,所以就用linux系统里面安装matlab运行环境来解决这个问题,我们仍然可以把人家写的matlab程序,在linux命令行下面,当做一个脚本来运行!
比如,这次我就需要用broadinstitute的一个软件:Mutsig,找cancer driver gene的,http://www.broadinstitute.org/cancer/cga/mutsig_run,但是我看了说明才发现,它是用matlab写的,所以我要想在我的服务器用,就必须按照安装matlab运行环境,在官网可以下载:http://www.mathworks.com/products/compiler/mcr/
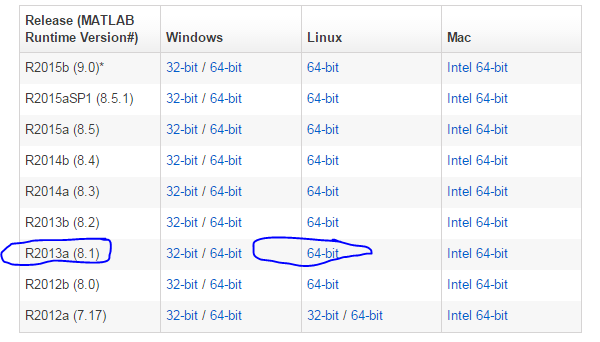
我这里选择的是R2013a (8.1),下载之后解压是这样的,压缩包约四百多M
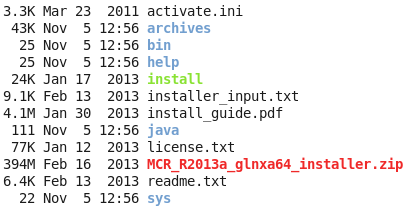
然后直接在解压后的目录里面运行那个install即可,然后如果你的linux可以传送图像,那么就会想安装windows软件一样方便!如果你的linux是纯粹的命令行,那么,就需要一步步的命令行交互,选择安装地址,等等来安装了。
记住你安装之后,会显示一些环境变量给你,请千万要记住,然后自己去修改自己的环境变量,如果你忘记了,就需要搜索来解决环境变量的问题啦!安装之后是这样的:
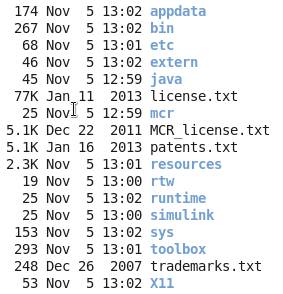
请记住你的安装目录,以后你运行其它matlab相关的程序,都需要把这个安装目录,当做一个参数传给你的其它程序的!!!
如果你没有设置环境变量,就会出各种各样的错误,用下面这个脚本可以设置
其中MCRROOT一般是$path/biosoft/matlab_running/v81/ 这样的东西,请务必注意,LD_LIBRARY_PATH非常重要,非常重要,非常重要!!!!
MCRROOT=$1
echo ---
LD_LIBRARY_PATH=.:${MCRROOT}/runtime/glnxa64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRROOT}/bin/glnxa64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRROOT}/sys/os/glnxa64;
MCRJRE=${MCRROOT}/sys/java/jre/glnxa64/jre/lib/amd64 ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/native_threads ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/server ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE}/client ;
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:${MCRJRE} ;
XAPPLRESDIR=${MCRROOT}/X11/app-defaults ;
export LD_LIBRARY_PATH;
export XAPPLRESDIR;
echo LD_LIBRARY_PATH is ${LD_LIBRARY_PATH};
如果你没有设置正确,那么会报一下的错误!
error while loading shared libraries: libmwmclmcrrt.so.8.1: cannot o pen shared object file: No such file or directory
error while loading shared libraries: libmwlaunchermain.so: cannot o pen shared object file: No such file or directory
等等!!!
竹子-博客(.NET/Java/Linux/架构/管理/敏捷)
思索、感悟、践行!走向高效,快乐,平衡!
已下目录是本人用爬虫爬取的!
每天一个linux命令(61):wget命令
每天一个linux命令(60):scp命令
每天一个linux命令(59):rcp命令
每天一个linux命令(58):telnet命令
每天一个linux命令(57):ss命令
每天一个linux命令(56):netstat命令
每天一个linux命令(55):traceroute命令
每天一个linux命令(54):ping命令
每天一个linux命令(53):route命令
每天一个linux命令(52):ifconfig命令
每天一个linux命令(51):lsof命令
每天一个linux命令(50):crontab命令
每天一个linux命令(49):at命令
每天一个linux命令(48):watch命令
每天一个linux命令(47):iostat命令
每天一个linux命令(46):vmstat命令
每天一个linux命令(45):free 命令
每天一个linux命令(44):top命令
每天一个linux命令(43):killall命令
每天一个linux命令(42):kill命令
每天一个linux命令(41):ps命令
每天一个linux命令(40):wc命令
每天一个linux命令(39):grep 命令
每天一个linux命令(38):cal 命令
每天一个linux命令(37):date命令
每天一个linux命令(36):diff 命令
每天一个linux命令(35):ln 命令
每天一个linux命令(34):du 命令
每天一个linux命令(33):df 命令
每天一个linux命令(32):gzip命令
每天一个linux命令(31): /etc/group文件详解
每天一个linux命令(30): chown命令
每天一个linux命令(29):chgrp命令
每天一个linux命令(28):tar命令
每天一个linux命令(27):linux chmod命令
每天一个linux命令(26):用SecureCRT来上传和下载文件
每天一个linux命令(25):linux文件属性详解
每天一个linux命令(24):Linux文件类型与扩展名
每天一个linux命令(23):Linux 目录结构
每天一个linux命令(22):find 命令的参数详解
每天一个linux命令(21):find命令之xargs
每天一个linux命令(20):find命令之exec
每天一个linux命令(19):find 命令概览
每天一个linux命令(18):locate 命令
每天一个linux命令(17):whereis 命令
每天一个linux命令(16):which命令
每天一个linux命令(15):tail 命令
每天一个linux命令(14):head 命令
每天一个linux命令(13):less 命令
每天一个linux命令(12):more命令
每天一个linux命令(11):nl命令
每天一个linux命令(10):cat 命令
每天一个linux命令(9):touch 命令
每天一个linux命令(8):cp 命令
每天一个linux命令(7):mv命令
每天一个linux命令(6):rmdir 命令
每天一个linux命令(5):rm 命令
每天一个linux命令(4):mkdir命令
每天一个linux命令(3):pwd命令
每天一个linux命令(2):cd命令
每天一个linux命令(1):ls命令
需要插件和自己修改主题下面的foot.php代码。
参考 http://jingyan.baidu.com/article/ae97a646ce37c2bbfd461d01.html
步骤如下:
1、登陆到wp后台,鼠标移动到左侧菜单的“插件”链接上,会弹出子菜单,点击子菜单的“安装插件”链接
2、WP-PostViews插件显示wordpress文章点击浏览量
在“安装插件”链接页面的搜索框中输入“WP-PostViews”,然后回车
3、WP-PostViews插件显示wordpress文章点击浏览量
在搜索结果页面点击“WP-PostViews”插件内容区域的“现在安装”按钮
4、WP-PostViews插件显示wordpress文章点击浏览量
程序自动下载插件到服务器并解压安装,一直等到安装成功信息出现,然后在安装成功提示页面点击“启动插件”链接。
5、WP-PostViews插件显示wordpress文章点击浏览量
页面会自动跳转到“已安装插件”页面,在已安装插件列表中我们可以看到“Form Manager”插件已经处于启用状态(插件名下是“停用”链接)。
有了这个插件之后,我们的整个网页环境里面就多了一个 the_views()函数,它统计着每个文章的点击数,这样我们之前的网页就能显示点击数了。
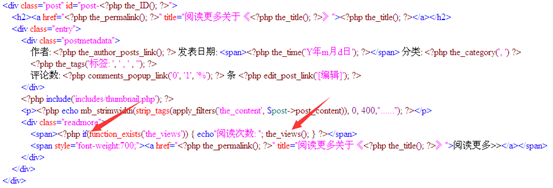
这个是我现在用的主题的php代码,把文章用span标记隔开了,而且显示着上面php代码里面的每一个内容包括日期,分类,标签,评论等等
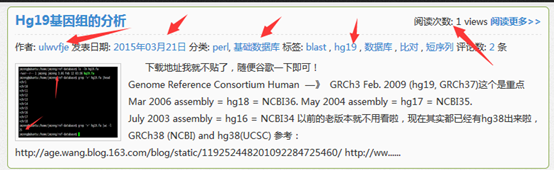
其中thez-view()这个函数返回的不仅仅是一个访客数,但是我的文章的访客都太少了,所以我写了一个脚本帮我刷一刷流量。
[perl]
use List::MoreUtils qw(uniq);
$page='http://www.bio-info-trainee.com/?paged=';
foreach (1..5){ #我的文章比较少,就42个,所以只有5个页面
$url_page=$page.$_;
$tmp=`curl $url_page`;
#@p=$tmp=~/p=(\d+)/;
$tmp =~ s/(p=\d+)/push @p, $1/eg; #寻找p=数字这样的标签组合成新的网页地址
}
@p=uniq @p;
print "$_\n" foreach @p; #可以找到所有42个网页的地址
foreach (@p){
$new_url='http://www.bio-info-trainee.com/?'.$_;
`curl $new_url` foreach (1..100); #每个网页刷一百次
}
[/perl]
大家可以看到这个网页被刷的过程,从15到21到27直到100
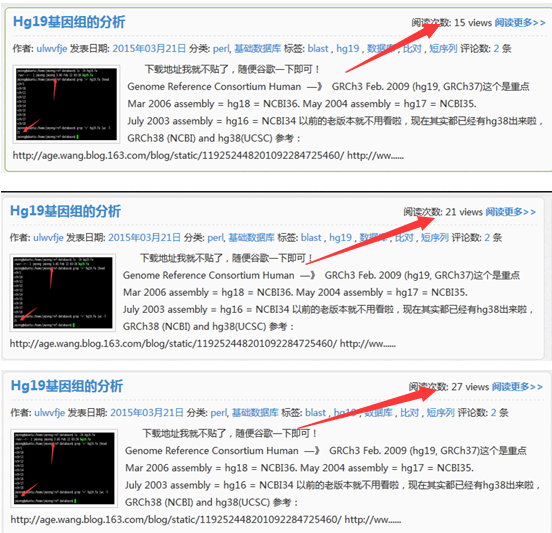
大家现在再去看我的网页,就每个文章都有一百的访问量啦!
http://www.bio-info-trainee.com/
想了想,既然是菜鸟教程,那就索性再介绍点更基础的东西,基本上只要是大学毕业的都能看懂,不需要懂计算机了。首先讲讲linux服务器吧,因为生物信息也算是半个大数据分析,所以我们平常的办公电脑一般都是不能满足需求的,大部分实验室及公司都会自己配置好服务器给菜鸟们用,菜鸟们首先要拿到服务器的IP和高手给你的用户名和密码。
一般我们讲服务器,大多是linux系统,而我这里所讲的linux系统呢,特指ubuntu,其余的我懒得管了,大家也不要耗费无谓的时间纠结那些名词的不同!
登录到服务器有两种方法,一种是ssh,传输你的命令给服务器执行,另一种是ftp,和服务器交换文件。而ssh我们通常用putty,xshell等等。ftp呢,我们可以用winscp,xshell,所以我一直都用xshell,因为它两者都能搞定!
Xshell软件自行搜索下载,打开之后新建一个连接,然后登陆即可。
然后输入以下命令,可以查看服务器配置,包括cpu。内存,还有硬盘
cat /proc/cpuinfo |grep pro|wc -l
free -g
df -h
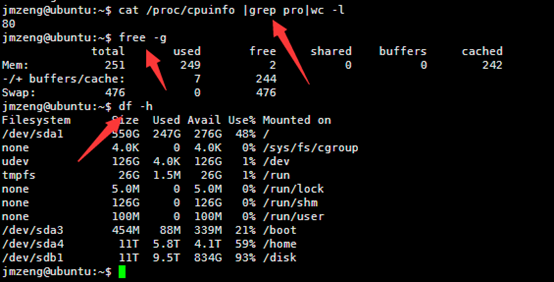
这个服务器配置好一点,有80个cpu,内存256G,硬盘有2个11T的,是比较成熟的配置。
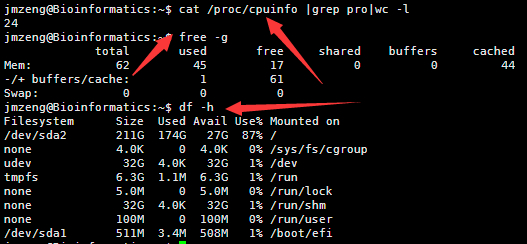
这个是一个小型服务器。也就24个核,64G的内存,但是存储量有点小呀,其实可以随便花几百块钱买个1T的硬盘挂载上去的。
然后linux的其它命令大家就得自己去搜索一个个使用,然后熟悉,记牢,然后创新啦!
我随便敲几个我常用的吧: ls cd mkdir rm cp cat head tail more less diff grep awk sed grep perl 等等!
呀,突然间发现我才介绍了ssh的方法登陆服务器并且发送命令在服务器上面运行,下面贴图如何传输文件。一般xshell的菜单里面有绿的文件夹形式的标签就是打开ftp文件传输,这种可视化的软件,大家慢慢摸索吧!
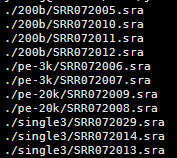
脚本类似于下面的样子,大家可以读懂之后就仿写
for i in *sra
do
echo $i
/home/jmzeng/bio-soft/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump --split-3 $i
Done
这个脚本是把当前目录下所有的NCBI下载的sra文件都加压开来成测序fastq格式文件
有这些数据,分布在不同的目录,如果是写命令一个个文件处理,很麻烦,如果有几百个那就更麻烦了,所以需要用shell脚本
这样只需要bash这个脚本即可一次性处理所有的数据
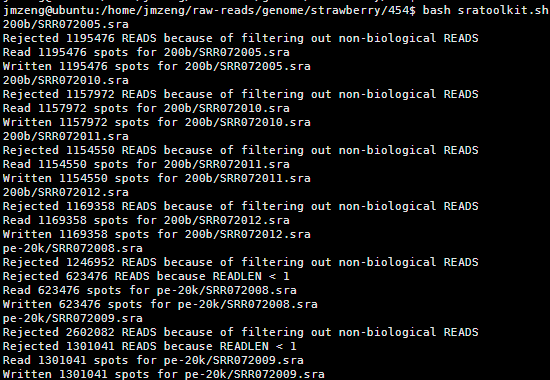
还有很多类似的脚本,非常简单的
for i in *fq
do
echo $i
bowtie2 -p 13 -x ../../RNA.fa -U $i -S $i.sam
done
for i in */accepted_hits.bam
do
echo $i
out=`echo $i |cut -d'/' -f 1`_clout
samtools mpileup -guSDf /home/immune/refer_genome/hg19/hg19.fa $i | bcftools view -cvNg - >snp-vcf/$out.vcf
done
while read id
do
echo $id
out=`echo $id |cut -d'/' -f 2`
reads=`echo $id |cut -d'/' -f 3|sed 's/\r//g'`
tophat2 -p 13 -o $out /home/immune/refer_genome/hg19/hg19 $reads
done <$1
等等
查某个基因家族在某物种的具体信息
我很伤心,不知道是不是我写的教程还是不够人性化,一个朋友在群里面问如何知道NAC基因家族在拟南芥里面的105个基因信息,我随便给他示范了一下在人类里面如何找,希望他能触类旁通,结果他不会linux,啥生信基础都没有,我只会诱导他简单学习一下,希望他至少明白什么的taxid。所以我给了他我之前写的教程,只希望他告诉我拟南芥的taxid我就帮他把那105个基因找出来。 Continue reading