一:安装并加装该R包
安装就用source("http://bioconductor.org/biocLite.R") ;biocLite("DESeq")即可,如果安装失败,就需要自己下载源码包,然后安装R模块。
二.所需要数据
它的说明书指定了我们一个数据
source("http://bioconductor.org/biocLite.R") ;biocLite("pasilla")
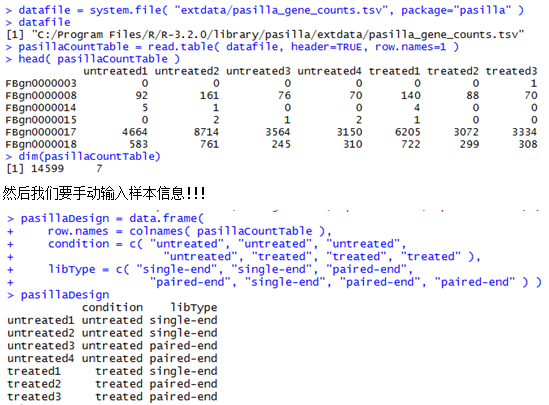
安装了pasilla这个包之后,在这个包的安装目录就可以找到一个表格文件,就是我们的DESeq需要的文件。
C:\Program Files\R\R-3.2.0\library\pasilla\extdata\pasilla_gene_counts.tsv
说明书原话是这样的
The table cell in the i-th row and the j-th column of the table tells how many reads have been mapped to gene i in sample j.
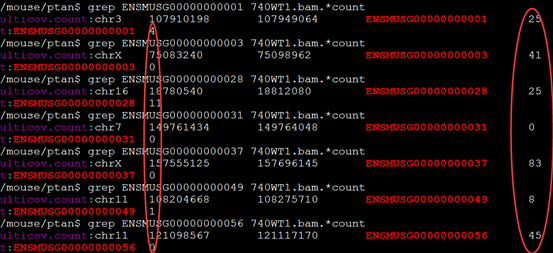
一般我们需要用htseq-count这个程序对我们的每个样本的sam文件做处理计数,并合并这样的数据
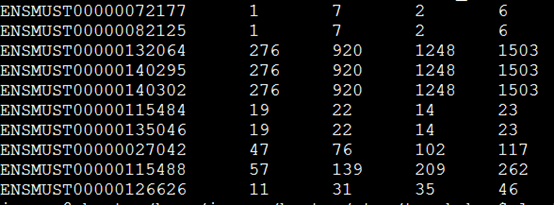
下面这个是示例数据,第一列是基因ID号,后面的每一列都是一个样本。
de = newCountDataSet( pasillaCountTable, condition ) #根据我们的样本因子把基因计数表格读入成一个cds对象,这个newCountDataSet函数就是为了构建对象!
对我们构建好的de对象就可以直接开始找差异啦!非常简单的几步即可
de=estimateSizeFactors(de)
de=estimateDispersions(de)
res=nbinomTest(de,"K","W") #最重要的就是这个res表格啦!
uniq=na.omit(res)
我这里是对4个样本用htseq计数后的文件来做的,贴出完整代码吧
library(DESeq)
#首先读取htseq对bam或者sam比对文件的计数结果
K1=read.table("742KO1.count",row.names=1)
K2=read.table("743KO2.count",row.names=1)
W1=read.table("740WT1.count",row.names=1)
W2=read.table("741WT2.count",row.names=1)
data=cbind(K1,K2,W1,W2)
data=data[-c(43630:43634),]
#把我们的多个样本计数结果合并起来成数据框,列是不同样本,行是不同基因
colnames(data)=c("K1","K2","W1","W2")
type=rep(c("K","W"),c(2,2))
#构造成DESeq的对象,并对分组样本进行基因表达量检验
de=newCountDataSet(data,type)
de=estimateSizeFactors(de)
de=estimateDispersions(de)
res=nbinomTest(de,"K","W")
#res就是我们的表达量检验结果
library(org.Mm.eg.db)
tmp=select(org.Mm.eg.db, keys=res$id, columns="GO", keytype="ENSEMBL")
ensembl_go=unlist(tapply(tmp[,2],as.factor(tmp[,1]),function(x) paste(x,collapse ="|"),simplify =F))
#首先输出所有的计数数据,加上go注释信息
all_res=res
res$go=ensembl_go[res$id]
write.csv(res,file="all_data.csv",row.names =F)
#然后输出有意义的数据,即剔除那些没有检测到表达的基因
uniq=na.omit(res)
sort_uniq=uniq[order(uniq$padj),]
write.csv(sort_uniq,file="sort_uniq.csv",row.names =F)
#然后挑选出padj值小于0.05的差异基因数据来做富集,富集用的YGC的两个包,在我前面的博客已经详细说明了!
tmp=select(org.Mm.eg.db, keys=sort_uniq[sort_uniq$padj<0.05,1], columns="ENTREZID", keytype="ENSEMBL")
diff_ENTREZID=tmp$ENTREZID
require(DOSE)
require(clusterProfiler)
diff_ENTREZID=na.omit(diff_ENTREZID)
ego <- enrichGO(gene=diff_ENTREZID,organism="mouse",ont="CC",pvalueCutoff=0.01,readable=TRUE)
ekk <- enrichKEGG(gene=diff_ENTREZID,organism="mouse",pvalueCutoff=0.01,readable=TRUE)
write.csv(summary(ekk),"KEGG-enrich.csv",row.names =F)
write.csv(summary(ego),"GO-enrich.csv",row.names =F)