在R语言里面的bioconductor系列包里面有一个物种注释信息包,其中人类是org.Hs.eg.db
里面有基于人的hg19基因组版本的大部分基因信息之间的转换数据!
包括基因的entrez ID,symbol,name,locus,refseq,kegg pathway,GO ontology对应关系!
但是它们散布在该包的各个数据结构里面!
> ls("package:org.Hs.eg.db")
[1] "org.Hs.eg" "org.Hs.eg.db"
[3] "org.Hs.eg_dbconn" "org.Hs.eg_dbfile"
[5] "org.Hs.eg_dbInfo" "org.Hs.eg_dbschema"
[7] "org.Hs.egACCNUM" "org.Hs.egACCNUM2EG"
[9] "org.Hs.egALIAS2EG" "org.Hs.egCHR"
[11] "org.Hs.egCHRLENGTHS" "org.Hs.egCHRLOC"
[13] "org.Hs.egCHRLOCEND" "org.Hs.egENSEMBL"
[15] "org.Hs.egENSEMBL2EG" "org.Hs.egENSEMBLPROT"
[17] "org.Hs.egENSEMBLPROT2EG" "org.Hs.egENSEMBLTRANS"
[19] "org.Hs.egENSEMBLTRANS2EG" "org.Hs.egENZYME"
[21] "org.Hs.egENZYME2EG" "org.Hs.egGENENAME"
[23] "org.Hs.egGO" "org.Hs.egGO2ALLEGS"
[25] "org.Hs.egGO2EG" "org.Hs.egMAP"
[27] "org.Hs.egMAP2EG" "org.Hs.egMAPCOUNTS"
[29] "org.Hs.egOMIM" "org.Hs.egOMIM2EG"
[31] "org.Hs.egORGANISM" "org.Hs.egPATH"
[33] "org.Hs.egPATH2EG" "org.Hs.egPFAM"
[35] "org.Hs.egPMID" "org.Hs.egPMID2EG"
[37] "org.Hs.egPROSITE" "org.Hs.egREFSEQ"
[39] "org.Hs.egREFSEQ2EG" "org.Hs.egSYMBOL"
[41] "org.Hs.egSYMBOL2EG" "org.Hs.egUCSCKG"
[43] "org.Hs.egUNIGENE" "org.Hs.egUNIGENE2EG"
[45] "org.Hs.egUNIPROT"
>
其实比较重要的信息,我们需要把它们统一关联起来,做成一个表格,这样可以上传到数据库里面,做成网页,可视化展现,查找!
suppressMessages(library(org.Hs.eg.db))
all_EG=mappedkeys(org.Hs.egSYMBOL)
###下面的表格最后是基于entrez ID而关联起来的!
tmp=unlist(as.list(org.Hs.egSYMBOL))
EG2Symbol=data.frame(EGID=names(tmp),symbol=as.character(tmp))
tmp=unlist(as.list(org.Hs.egENSEMBL))
EG2ENSEMBL=data.frame(EGID=names(tmp),ENSEMBL=as.character(tmp))
tmp=unlist(as.list(org.Hs.egGENENAME))
EG2name=data.frame(EGID=names(tmp),name=as.character(tmp))
tmp=unlist(as.list(org.Hs.egMAP))
EG2MAP=data.frame(EGID=names(tmp),MAP=as.character(tmp))
###EG2GO and using mySQL
EG2path=as.list(org.Hs.egPATH)
EG2path=lapply(EG2path, function(x) paste(x,collapse = ":"))
tmp=unlist(EG2path)
EG2path=data.frame(EGID=names(tmp),path=as.character(tmp))
#as.list(head(org.Hs.egGO2ALLEGS))
GO2allEG=as.list(org.Hs.egGO2ALLEGS)
tmp=unlist(GO2allEG)
names(tmp)=substring(names(tmp),1,10) ## change GO:0000002.IMP to GO:0000002
EG2allGO <- tapply(tmp,tmp,function(x){names(x)})
EG2allGO=lapply(EG2allGO, function(x) paste(x,collapse = ":"))
tmp=unlist(EG2allGO)
EG2GO=data.frame(EGID=names(tmp),GO=as.character(tmp))
tmp=merge(EG2Symbol,EG2MAP,by='EGID',all=TRUE)
tmp=merge(tmp,EG2ENSEMBL,by='EGID',all=TRUE)
tmp=merge(tmp,EG2path,by='EGID',all=TRUE)
tmp=merge(tmp,EG2name,by='EGID',all=TRUE)
my_gene_mapping=merge(tmp,EG2GO,by='EGID',all=TRUE)
##[1] "EGID" "symbol" "MAP" "ENSEMBL" "path" "name" "GO"
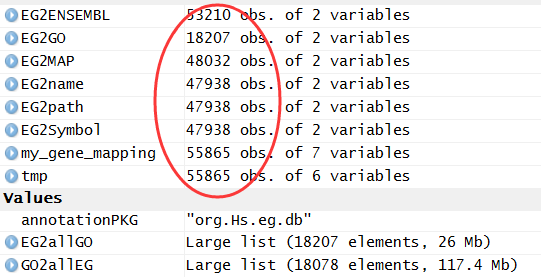
因为我用的merge的all=TRUE,所以最后记录会越来越多
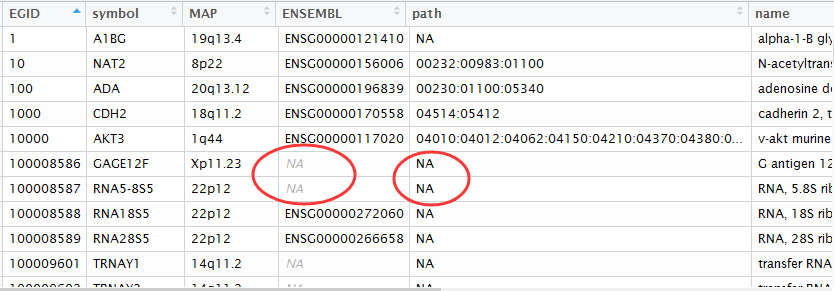
最后的结果就是下面这样,各种ID转换都储存到了my_gene_mapping这个数据对象里面!
可以看到有些基因是没有ensemble数据库对应的ID的,非常多的基因没有被注释到KEGG通路数据库!
我这里如果一个基因对应多个通路,我用冒号连接起来了,成了一个字符串!在后面会有用!
现在需要把它上传到数据库里面,这样我就可以用R的shiny可视化网页来查找数据库,做ID转换啦!!!
suppressMessages(library(RMySQL))
con <- dbConnect(MySQL(), host="127.0.0.1", port=3306, user="root", password="11111111")
dbSendQuery(con, "USE test")
dbWriteTable(con,'my_gene_mapping',my_gene_mapping)
dbDisconnect(con)
然后就可以在自己的mysql里面看到它啦!!!
其实 "org.Hs.egOMIM" 也很重要,不过我这里只是举个例子,就不深究了!
还有一点,就是那个pathway ID,因为我要以entrez ID为主键,所以把多个pathway给合并了!
suppressMessages(library(org.Hs.eg.db))
kegg2ID=toTable(org.Hs.egPATH)
#[1] "gene_id" "path_id"
dbWriteTable(con,'kegg2ID_bioconductor',kegg2ID,row.name=F,overwrite=T)
go2id=toTable(org.Hs.egGO2ALLEGS)
## gene_id go_id Evidence Ontology
dbWriteTable(con,'go2id_bioconductor',go2id,row.name=F,overwrite=T)
dbDisconnect(con)
用这个代码,可以在数据库里面多创建两个表,会有用的!