有这个需求,是因为我们经常对某些细胞系进行有针对性的设计变异,比如BAF155的R1064K呀,H3F3A的K27呀,那我我们拿到高通量测序数据的时候,就肯定希望可以快速的看看这个基因是否被突变成功了。现在比对几乎不耗费什么时间了,但是得到的sam要sort的时候还是蛮耗费时间的。假设,我们已经得到了所有样本的sort好的bam文件,想看看自己设计的基因突变是否成功了,可以有针对性的只call 某个基因的突变!
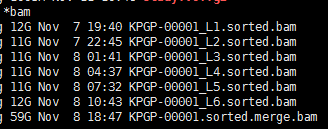
有这个需求,是因为我们经常对某些细胞系进行有针对性的设计变异,比如BAF155的R1064K呀,H3F3A的K27呀,那我我们拿到高通量测序数据的时候,就肯定希望可以快速的看看这个基因是否被突变成功了。现在比对几乎不耗费什么时间了,但是得到的sam要sort的时候还是蛮耗费时间的。假设,我们已经得到了所有样本的sort好的bam文件,想看看自己设计的基因突变是否成功了,可以有针对性的只call 某个基因的突变!
我对H3F3A这个基因做了两个突变的cellline,分别是G34V和K27M,现在知道这个基因在hg38上面的坐标是:
Genomic Location for H3F3A Gene
Chromosome: 1
Start:226,061,851 bp from pter End:226,072,002 bp from pter
Size:10,152 bases Orientation:Plus strand
然后我用samtools结合bcftools把该基因区域的snp位点call出来:
samtools mpileup -r chr1:226061851-226072001 -t "DP4" -ugf ~/reference/genome/hg38/hg38.fa *sorted.bam | bcftools call -vmO z -o H3F3A.vcf.gz
产品特点:
1.涵盖超过1,800,000个遗传变异标志物:包括超过906,600个SNP和超过946,000个用于检测拷贝数变化(CNV,Copy Number Variation)的探针;
2.SNP和CNV两种探针高密度且均匀地分布在整个基因组,不仅可以用于SNP基因精确分型,还可用于拷贝数变异CNV的研究;
3.744,000个探针平均分配到整个基因组上,用来发现未知的拷贝数变异区域;
4.可用于Copy-neutral LOH/UPD检测,亲子鉴定,纯合性分析、血缘关系鉴定、遗传病或其它疾病的研究。

其中freebayes,bcftools,gatk都是把所有的snp细节都call出来了,可以看到下面这些软件的结果有的高达一百多万个snp,而一般文献都说外显子组测序可鉴定约8万个变异!
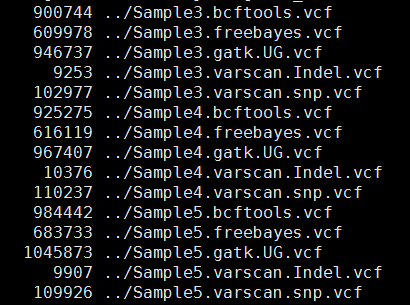
这样得到突变太多了,所以需要过滤。这里过滤的统一标准都是qual大于20,测序深度大于10。过滤之后的snp数量如下
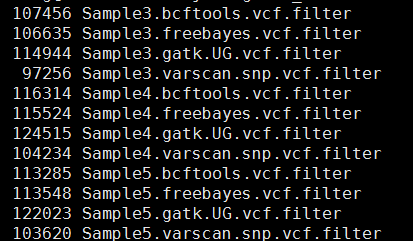
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample3.bcftools.vcf >Sample3.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample4.bcftools.vcf >Sample4.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample5.bcftools.vcf >Sample5.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample3.freebayes.vcf > Sample3.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample4.freebayes.vcf > Sample4.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample5.freebayes.vcf > Sample5.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample3.gatk.UG.vcf >Sample3.gatk.UG.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample4.gatk.UG.vcf >Sample4.gatk.UG.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample5.gatk.UG.vcf >Sample5.gatk.UG.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample3.varscan.snp.vcf >Sample3.varscan.snp.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample4.varscan.snp.vcf >Sample4.varscan.snp.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample5.varscan.snp.vcf >Sample5.varscan.snp.vcf.filter
这样不同工具产生的snp记录数就比较整齐了,我们先比较四种不同工具的call snp的情况,然后再比较三个人的区别。
然后写了一个程序把所有的snp合并起来比较
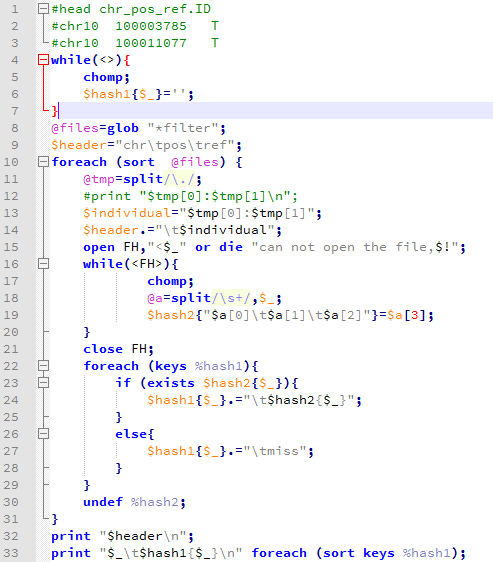
得到了一个很有趣的表格,我放在excel里面看了看 ,主要是要看生物学意义,但是我的生物学知识好多都忘了,得重新学习了 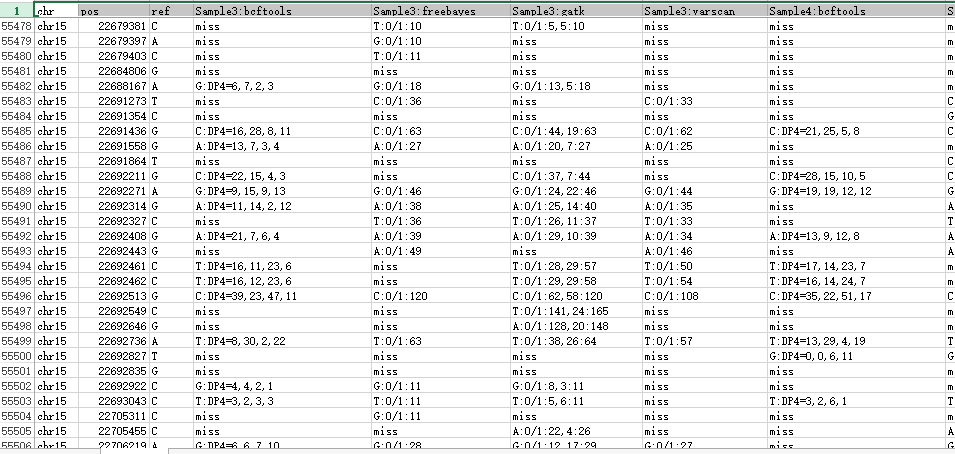
准备文件:下载必备的软件和参考基因组数据
1、软件
ps:还有samtools,freebayes和varscan软件,我以前下载过,这次就没有再弄了,但是下面会用到
2、参考基因组
3、参考 突变数据
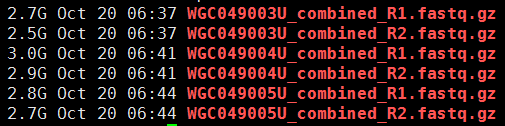
第一步,下载数据
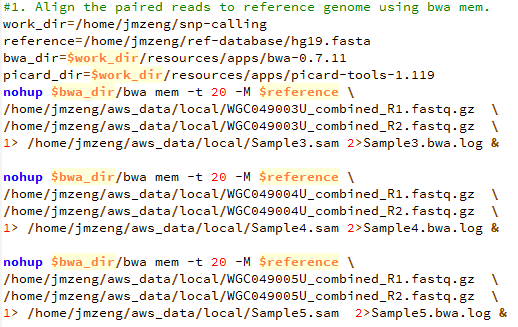
第二步,bwa比对
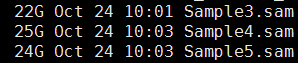
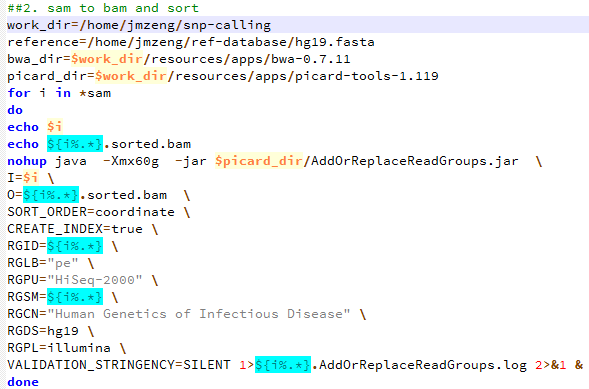
第三步,sam转为bam,并sort好
第四步,标记PCR重复,并去除
第五步,产生需要重排的坐标记录
第六步,根据重排记录文件把比对结果重新比对
第七步,把最终的bam文件转为mpileup文件
第八步,用bcftools 来call snp
第九步,用freebayes来call snp
第十步,用gatk 来call snp
第十一步,用varscan来call snp
下面的图片是按照顺序来的,我就不整理了

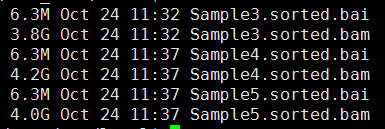
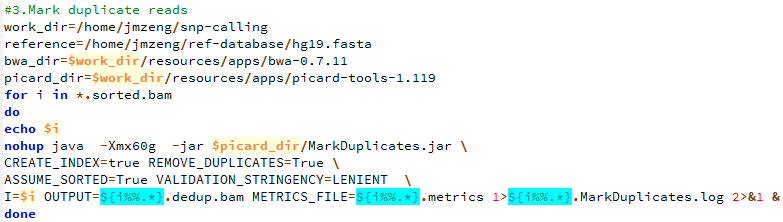
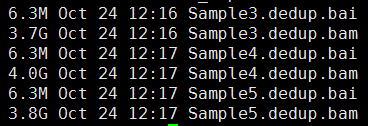
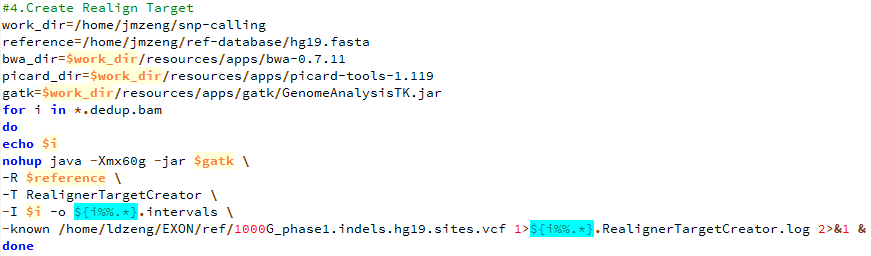
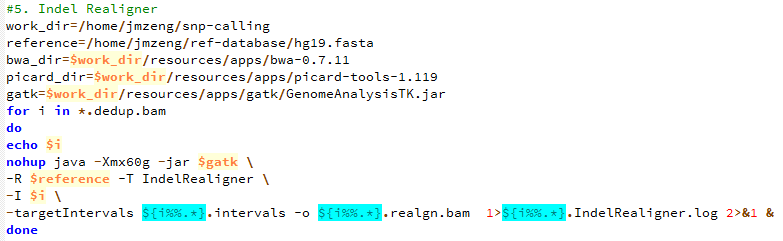
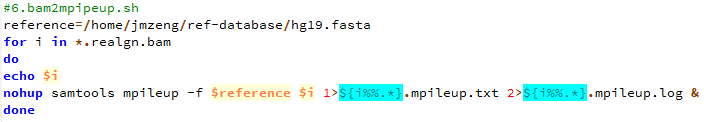
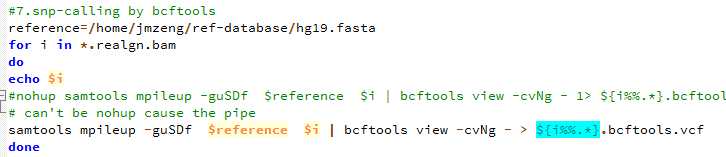
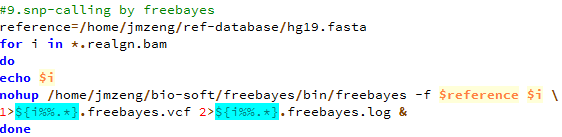
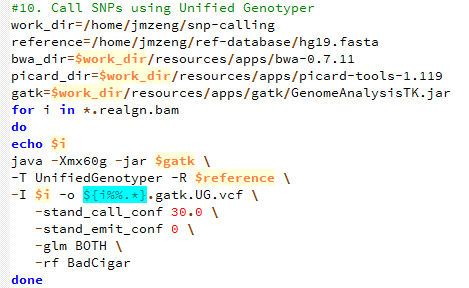
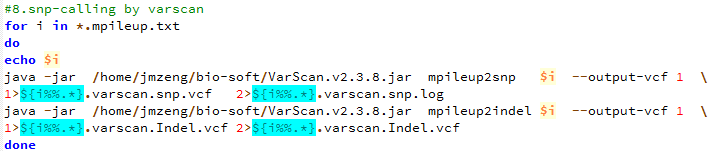
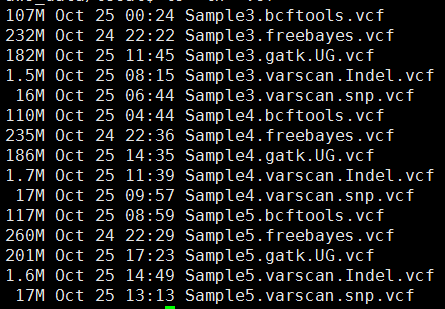
感觉最近接触的生物信息学知识越多,越对大数据时代的到来更有同感了。现在的研究者,其实很多都可以自己在家里做了,大量的数据基本都是公开的, 但是一个人闭门造车成就真的有限,与他人交流的思想碰撞还是蛮重要的。
我以前也是用这样的流程 SNP Pipeline Commands 1. Index the reference genome using bwa index /software/bwa-0.7.10/bwa index /reference/japonica/reference.fa 2. Align the paired reads to reference genome using bwa mem. Note: Specify the number of threads or processes to use using the -t parameter. The possible number of threads depends on the machine where the command will run. /software/bwa-0.7.10/bwa mem -M -t 8 /reference/japonica/reference.fa /reads/filename_1.fq.gz /reads/filename_2.fq.gz > /output/filename.sam 3. Sort SAM file and output as BAM file java -Xmx8g -jar /software/picard-tools-1.119/SortSam.jar INPUT=/output/filename.sam OUTPUT=/output/filename.sorted.bam VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE 4. Fix mate information java -Xmx8g -jar /software/picard-tools-1.119/FixMateInformation.jar INPUT=/output/filename.sorted.bam OUTPUT=/output/filename.fxmt.bam SO=coordinate VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE 5. Mark duplicate reads java -Xmx8g -jar /software/picard-tools-1.119/MarkDuplicates.jar INPUT=/output/filename.fxmt.bam OUTPUT=/output/filename.mkdup.bam METRICS_FILE=/output/filename.metrics VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=TRUE MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000 6. Add or replace read groups java -Xmx8g -jar /software/picard-tools-1.119/AddOrReplaceReadGroups.jar INPUT=/output/filename.mkdup.bam OUTPUT=/output/filename.addrep.bam RGID=readname PL=Illumina SM=readname CN=BGI VALIDATION_STRINGENCY=LENIENT SO=coordinate CREATE_INDEX=TRUE 7. Create index and dictionary for reference genome /software/samtools-1.0/samtools faidx /reference/japonica/reference.fa java -Xmx8g -jar /software/picard-tools-1.119/CreateSequenceDictionary.jar REFERENCE=/reference/japonica/reference.fa OUTPUT=/reference/reference.dict 8. Realign Target java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T RealignerTargetCreator -I /output/filename.addrep.bam -R /reference/japonica/reference.fa -o /output/filename.intervals -fixMisencodedQuals -nt 8 9. Indel Realigner java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T IndelRealigner -fixMisencodedQuals -I /output/filename.addrep.bam -R /reference/japonica/reference.fa -targetIntervals /output/filename.intervals -o /output/filename.realn.bam 10. Merge individual BAM files if there are multiple read pairs per sample /software/samtools-1.0/samtools merge /output/filename.merged.bam /output/*.realn.bam 11. Call SNPs using Unified Genotyper java -Xmx8g -jar /software/GenomeAnalysisTK-3.2-2/GenomeAnalysisTK.jar -T UnifiedGenotyper -R /reference/japonica/reference.fa -I /output/filename.merged.bam -o filename.merged.vcf -glm BOTH -mbq 20 --genotyping_mode DISCOVERY -out_mode EMIT_ALL_SITES
wget http://clavius.bc.edu/~erik/freebayes/freebayes-5d5b8ac0.tar.gz
tar xzvf freebayes-5d5b8ac0.tar.gz
cd freebayes
make
一个小插曲,安装的过程报错:/bin/sh: 1: cmake: not found
所以我需要自己下载安装cmake,然后把cmake添加到环境变量
首先下载源码包http://www.cmake.org/cmake/resources/software.html
wget http://cmake.org/files/v3.3/cmake-3.3.2.tar.gz
解压进去,然后源码安装三部曲,首先 ./configu 然后make 最后make install
cmake 会默认安装在 /usr/local/bin 下面,这里需要修改,因为你可能没有 /usr/local/bin 权限,安装到自己的目录,然后把它添加到环境变量!
step2:准备数据
wget http://bioinformatics.bc.edu/marthlab/download/gkno-cshl-2013/chr20.fawget ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/data/NA12878/alignment/NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam
wget ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/data/NA12878/alignment/NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam.bai用这个代码就可以下载千人基因组计划的NA12878样本的第20号染色体相关数据啦
freebayes -f chr20.fa \
NA12878.chrom20.ILLUMINA.bwa.CEU.low_coverage.20121211.bam >NA12878.chr20.freebayes.vcf一般就用默认参数即可
GATK这个软件在做snp-calling的时候使用率非常高,因为之前一直是简单粗略的看看snp情况而已,所以没有具体研究它。
这些天做一些外显子项目以找snp为重点,所以想了想还是用起它,报错非常多,调试了好久才成功。
所以记录一些注意事项!
GATK软件本身是受版权保护的,所以需要申请才能下载使用,大家自己去broad institute申请即可。
下载软件就可以直接使用,java软件不需要安装,但是需要你的机器上面有java,当然软件只是个开始,重点是你还得下载很多配套数据,https://software.broadinstitute.org/gatk/download/bundle(ps:这个链接可能会失效,下面的文件,请自己谷歌找到地址哈。),而且这个时候要明确你的参考基因组版本了!!! b36/b37/hg18/hg19/hg38,记住b37和hg19并不是完全一样的,有些微区别哦!!!
Continue reading
| 去除PCR重复前 | 样本名 | 去除PCR重复后 |
| 106082 | BC1-1.snp | 103829 |
| 101443 | BC1-2.snp | 99500 |
| 103937 | BC2-1.snp | 101833 |
| 102979 | BC2-2.snp | 101022 |
| 105876 | BC3-1.snp | 103562 |
| 109168 | BC3-2.snp | 107052 |
| 107155 | BC4-1.snp | 104894 |
| 108335 | BC4-2.snp | 106031 |
| 100236 | BC5-1.snp | 98417 |
| 102322 | BC5-2.snp | 100395 |
| 103466 | BC6-1.snp | 101405 |
| 112940 | BC6-2.snp | 110611 |
| 113166 | BC7-1.snp | 110948 |
| 114038 | BC7-2.snp | 116090 |
| 123670 | PC1-1.snp | 121697 |
| 111402 | PC1-2.snp | 109389 |
| 106917 | PC2-1.snp | 105149 |
| 108724 | PC2-2.snp | 106776 |
可以看到去除pcr重复这个脚本对snp-calling的结果影响甚小,就是少了那么一千多个snp,脚本如下,我是用picard-tools进行的去除PCR重复,当然也可以用samtools来进行同样的步骤
[shell]
<b>for i in *.sorted.bam</b>
<b>do</b>
<b>echo $i</b>
<b>java -Xmx120g -jar /home/jmzeng/snp-calling/resources/apps/picard-tools-1.119/MarkDuplicates.jar \</b>
<b>CREATE_INDEX=true REMOVE_DUPLICATES=True \</b>
<b>ASSUME_SORTED=True VALIDATION_STRINGENCY=LENIENT METRICS_FILE=/dev/null \</b>
<b>INPUT=$i OUTPUT=${i%%.*}.sort.dedup.bam</b>
<b>done</b>
[/shell]
然后我们首先看看没有产生变化的那些snp信息的改变
head -50 ../rmdup/out/snp/BC1-1.snp |tail |cut -f 1,2,8
chr1 17222 ADP=428;WT=0;HET=1;HOM=0;NC=0
chr1 17999 ADP=185;WT=0;HET=1;HOM=0;NC=0
chr1 18091 ADP=147;WT=0;HET=1;HOM=0;NC=0
chr1 18200 ADP=278;WT=0;HET=1;HOM=0;NC=0
chr1 24786 ADP=238;WT=0;HET=1;HOM=0;NC=0
chr1 25072 ADP=24;WT=0;HET=1;HOM=0;NC=0
chr1 29256 ADP=44;WT=0;HET=1;HOM=0;NC=0
chr1 29265 ADP=44;WT=0;HET=1;HOM=0;NC=0
chr1 29790 ADP=351;WT=0;HET=1;HOM=0;NC=0
chr1 29939 ADP=109;WT=0;HET=1;HOM=0;NC=0
head -50 BC1-1.snp |tail |cut -f 1,2,8
chr1 17222 ADP=457;WT=0;HET=1;HOM=0;NC=0
chr1 17999 ADP=196;WT=0;HET=1;HOM=0;NC=0
chr1 18091 ADP=155;WT=0;HET=1;HOM=0;NC=0
chr1 18200 ADP=313;WT=0;HET=1;HOM=0;NC=0
chr1 24786 ADP=254;WT=0;HET=1;HOM=0;NC=0
chr1 25072 ADP=25;WT=0;HET=1;HOM=0;NC=0
chr1 29256 ADP=46;WT=0;HET=1;HOM=0;NC=0
chr1 29265 ADP=46;WT=0;HET=1;HOM=0;NC=0
chr1 29790 ADP=440;WT=0;HET=1;HOM=0;NC=0
chr1 29939 ADP=123;WT=0;HET=1;HOM=0;NC=0
可以看到,同一位点的snp仍然可以找到,仅仅是对测序深度产生了影响
然后我们再看看去除PCR重复这个步骤减少了的snp,在原snp里面是怎么样的
perl -alne '{$file++ if eof(ARGV);unless ($file){$hash{"$F[0]_$F[1]"}=1} else {print if not exists $hash{"$F[0]_$F[1]"} } }' ../rmdup/out/snp/BC1-1.snp BC1-1.snp |less
这个脚本就可以把去除PCR重复找到的snp位点在没有去除PCR重复的找到的snp文件里面过滤掉,查看那些去除PCR重复之前独有的snp
Min. 1st Qu. Median Mean 3rd Qu. Max.
8.00 8.00 11.00 44.26 25.00 7966.00
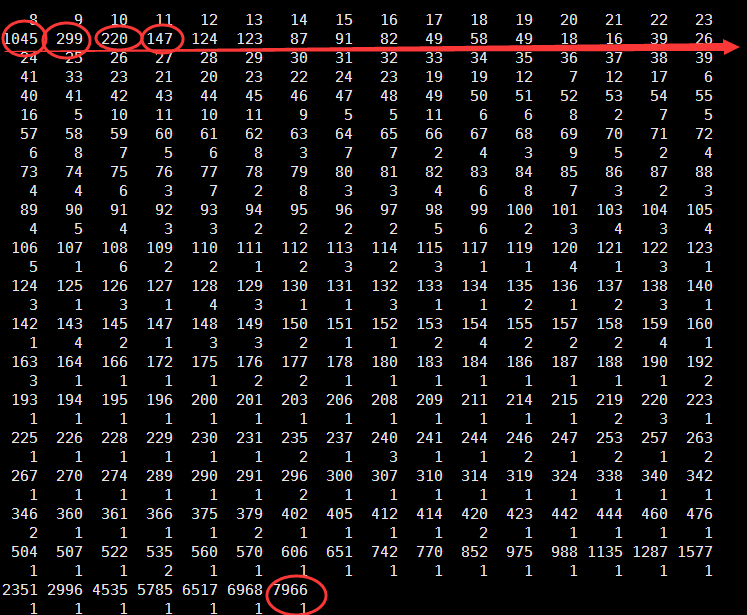
可以看到被过滤的snp大多都是测序深度太低了的,如下面的例子
chr1 726325 a 9 CCC.ccc,^:, IEHGHHG/9
chr1 726325 a 5 C.c,^:, IGH/9
chr1 726338 g 16 TTT.ttt,,....,,, IHGI:9<HIIFIHC5H
chr1 726338 g 10 T.t,,...,, II:HIIFH5H
可以看到这一步还是很有用的,但是怎么说呢,因为最后对snp的过滤本来就包含了一个步骤是对snp的测序深度小于20的给过滤掉
但是也有个别的测序深度非常高的snp居然也是被去除PCR重复这个步骤给搞没了!很奇怪,我还在探索之中.
grep 13777 BC1-1.mpileup |head
chr1 13777 G 263 ........,.C,,,,,.,,,.......,,,..,....,,......,.....c,........,,,,,,,..,...,,,,,.........,......C.......,,,,,,,,,,.....,,,,,,,.,,,..C,,,,,,CC,c,,,...C..,,,,cC.C..CC.CC,,cc,.C...C,,,,CCc,c,,,,,,,c,C.C.CC...C.cc,c...,C.CCcc...,CCC.C.CC..CCC..CC.c,cc,cc,,cc,C.,,^!.^6.^6.^6.^!, HIHIIIIEIEIHGIIIFIHIG?IIIIHIIHIFHIIHICIIIHIIGIEIIGIIIGHIIIIIIHIIHIHIIIIIIIHII1I?GHHHEHHIIEIEHIIEIIHHIIFIIIFHIHIIIIHIHIIHIIHHIIEIIIIIIHIIIIIIIIIG1HIIIIHIHIEHIHIHIIIIIIIIIIIHICIHIIIIIEIIIIHICIHGGIIIIIIIIEHIHIIIIIIHFIGGIIIIGIIIGICIIIHIIIIIIIIIIIHHHIIIIIHIIHDDII>>>>>
grep 13777 BC1-1.rmdup.mpileup |head
chr1 13777 G 240 ........,.C,,,,,.,,,.......,,,..,....,,......,....c,......,,,,,,,..,...,,,,,.........,......C......,,,,,,,,,,.....,,,,,,,.,,,..C,,,,,,CC,c,,,...C..,,,,cC..CC.CC,cc,.C...C,,,,CCc,c,,,,,,,cC.C.C..C.c,c...,C.CCcc...,CC.C.CCC..C.c,cc,,c,.,,^!.^6.^6.^!, HIHIIIIEIEIHGIIIFIHIG?IIIIHIIHIFHIIHICIIIHIIGIEIIIIIHIIIIIHIIHIHIIIIIIIHII1I?GHHHEHHIIEIEHIIEIHHIIFIIIFHIHIIIIHIHIIHIIHHIIEIIIIIIHIIIIIIIIIG1HIIIIHIHIEHIHIIIIIIIIIIHICIHIIIIIEIIIIHICIHGGIIIIIIHIHIIIIIHFIGGIIIIGIIIGCIIIIIIIIIIHHIIIHIHDII>>>>
然后我再搜索了一些
chr8 43092928 . A T . PASS ADP=7966;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:7967:7966:6261:1663:20.9%:0E0:39:39:3647:2614:1224:439
chr8 43092908 . T C . PASS ADP=6968;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:7002:6968:5315:1537:22.06%:0E0:37:38:3022:2293:890:647
chr8 43092898 . T G . PASS ADP=6517;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:6517:6517:4580:1587:24.35%:0E0:38:38:2533:2047:920:667
chr7 100642950 . T C . PASS ADP=770;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:771:770:615:155:20.13%:3.9035E-51:38:38:277:338:65:90
终于发现规律啦!!!原来它们的突变率都略高于20%,在没有去处PCR重复之前,是高于snp的阈值的,但是去除PCR重复对该位点的突变率产生了影响,使之未能通过筛选。
I'm using samtools mpileup and would like to generate both a pileup file and a vcf file as output. I can see how to generate one or the other, but not both (unless I run mpileup twice). I suspect I am missing something simple.
Specifically, calling mpileup with the -g or -u flag causes it to compute genotype likelihoods and output a bcf. Leaving these flags off just gives a pileup. Is there any way to get both, without redoing the work of producing the pileup file? Can I get samtools to generate the bcf _from_ the pileup file in some way? Generating the bcf from the bam file, when I already have the pileup, seems wasteful.
Thanks for any help!
我写了脚本来运行,才发现我居然需要两个重复的步骤来得到mpileup格式的数据和bcftools格式的数据,而这很明显的重复并且浪费时间的工作
for i in *sam
do
echo $i
samtools view -bS $i >${i%.*}.bam
samtools sort ${i%.*}.bam ${i%.*}.sorted
samtools index ${i%.*}.sorted.bam
samtools mpileup -f /home/jmzeng/ref-database/hg19.fa ${i%.*}.sorted.bam >${i%.*}.mpileup
samtools mpileup -guSDf /home/jmzeng/ref-database/hg19.fa ${i%.*}.sorted.bam | bcftools view -cvNg - > ${i%.*}.vcf
Done
我想得到mpileup格式,是因为后续的varscan等软件需要这个文件来call snp
而得到bcftools格式可以直接用bcftools进行snp-calling
samtools mpileup 命令只有用了-g或者-u那么就只会输出bcf文件
如果想得到mpileup格式的数据,就只能用-f参数。
至于如何安装该软件,请见上一个教程
一.首先把snp-calling步骤的VCF文件转为annovar软件要求的格式
convert2annovar.pl -format vcf4 12.vcf >12.annovar
二.进行注释
命令行参数比较多,还是用脚本来运行
# define path
infolder=/home/jmzeng/hoston/diff
outfolder=$infolder
annovardb=/home/jmzeng/bio-soft/annovar/humandb
# start annotating
/home/jmzeng/bio-soft/annovar/annotate_variation.pl \
--buildver hg19 \
--geneanno \
--outfile ${outfolder}/12.anno \
${infolder}/12.annovar \
${annovardb}
三.输出结果解读
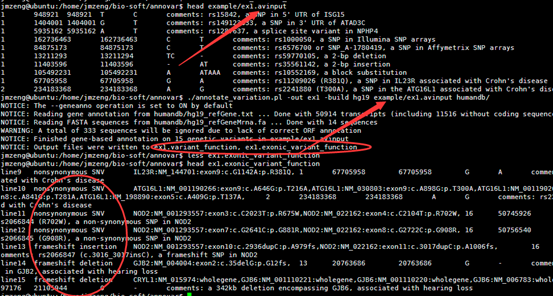
2.6M Apr 14 22:32 12.anno.exonic_variant_function
1.9K Apr 14 22:32 12.anno.log
1.3M Apr 14 22:32 12.anno.variant_function
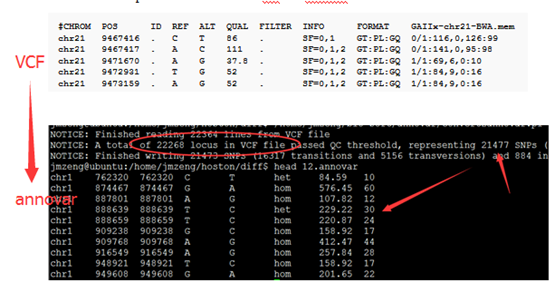
重点是后缀为exonic_variant_function,这个文件对每一个vcf的突变都进行了注释。
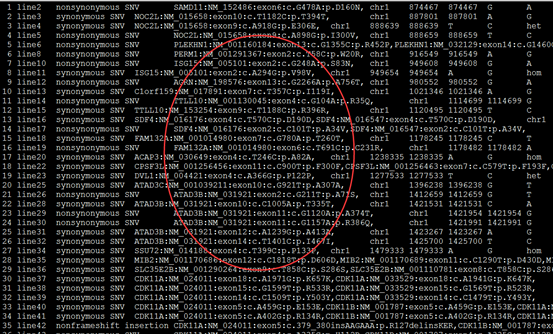
这个结果就可以用来解析了,可以根据实验设计来找到自己感兴趣的突变。
第5.6列是染色体及pos坐标
第4列信息非常复杂,是突变的注释
第12列是测序深度,一般要大于20
我这里是先把注释文件转换成以下格式
location:chr1:874467 SAMD11:NM_152486:exon6:c.G478A:p.D160N
location:chr1:888639 NOC2L:NM_015658:exon9:c.A918G:p.E306E
location:chr1:888659 NOC2L:NM_015658:exon9:c.A898G:p.I300V
location:chr1:916549 PERM1:NM_001291367:exon2:c.T58C:p.W20R
location:chr1:949608 ISG15:NM_005101:exon2:c.G248A:p.S83N
location:chr1:980552 AGRN:NM_198576:exon13:c.G2266A:p.A756T
location:chr1:1114699 TTLL10:NM_001130045:exon4:c.G104A:p.R35Q
location:chr1:1158631 SDF4:NM_016176:exon4:c.T570C:p.D190D
location:chr1:1158631 SDF4:NM_016547:exon4:c.T570C:p.D190D
location:chr1:1164073 SDF4:NM_016176:exon2:c.C101T:p.A34V
然后比较两个文件,取不同的突变来格式化输出。
我在NCBI里面下载了一个dbsnp_142数据库文件,发现它居然有2.5G的大小,我感到很不可思议,毕竟人的基因组也就3G,就30亿的碱基嘛。研究过的突然竟然有110,917,213 ,高达一亿个!!!
谁能给我解释一下呢!
而且人只有十万多个蛋白,2.2万多个基因!
jmzeng@ubuntu:/home/jmzeng/hoston/diff/snp$ wc -l dbsnp_142_chrom_id_rs
110917213 dbsnp_142_chrom_id_rs
jmzeng@ubuntu:/home/jmzeng/hoston/diff/snp$ tail dbsnp_142_chrom_id_rs
MT 16429 rs150751410
MT 16443 rs371960162
MT 16456 rs142662828
MT 16482 rs386419986
MT 16497 rs376846509
MT 16512 rs373943637
MT 16519 rs3937033
MT 16526 rs386829315
MT 16527 rs386829316
MT 16529 rs370705831
jmzeng@ubuntu:/home/jmzeng/hoston/diff/snp$ head dbsnp_142_chrom_id_rs
1 10108 rs62651026
1 10109 rs376007522
1 10139 rs368469931
1 10144 rs144773400
1 10150 rs371194064
1 10177 rs201752861
1 10177 rs367896724
1 10180 rs201694901
1 10228 rs143255646
1 10228 rs200462216
VCF是1000genome计划定义的测序比对突变说明文件,熟悉VCF文件的都知道,第三列是ID号,也就是说对该突变在dbsnp的数据库的编号。大多时候都是用点号占位,代表不知道在dbsnp的数据库的编号,这时候就需要我们自己来注释了。
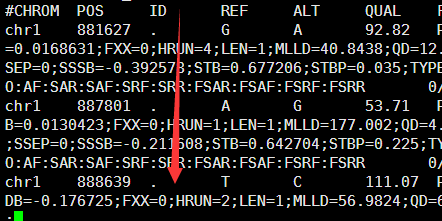
其实,这是一个非常简单的事情,因为有了CHROM和pos,只要找到一个文件,就可以自己写脚本来映射到它的ID号,但是找这个文件比较困难,我也是搜索了好久才找到的。
http://varianttools.sourceforge.net/Annotation/DbSNP
这里面提到了最新版的数据库是dbSNP138
The default version of our dbSNP annotation is currently referring to dbSNP138 (using hg19 coordinates) as shown below. However, users can also retrieve older versions of dbSNP: db135, dbSNP129, dbSNP130, dbSNP131 and dbSNP132. The 129 and 130 versions use hg18 as a reference genome and 131, 132, 135 and later use hg19. The archived versions can be used by a variant tools project by referring to their specific names - for example: dbSNP-hg18_129.
所以我就换了关键词,终于搜的了
http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?view+summary=view+summary&build_id=138
http://asia.ensembl.org/info/genome/variation/sources_documentation.html?redirect=no
SNP 138 database (232,952,851 million altogether).

有一个bioconductor包是专门来做snp过滤的
http://www.bioconductor.org/packages/release/bioc/html/VariantAnnotation.html
首先下载vcf文件。
nohup wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/00-All.vcf.gz &
这个文件很大,解压开是
如果大家对snp不了解,可以去查看它的各种介绍以及分类
http://moma.ki.au.dk/genome-mirror/cgi-bin/hgTrackUi?db=hg19&g=snp138
其实我这里本来有个hg19_snp141.txt文件,如下
1 10020 A - .
1 10108 C T .
1 10109 A T .
1 10139 A T .
1 10145 A - .
1 10147 C - .
1 10150 C T .
1 10177 A C .
1 10180 T C .
1 10229 A - .
还可以下载一些文件,如bed_chr_1.bed
chr1 175292542 175292543 rs171 0 -
chr1 20542967 20542968 rs242 0 +
chr1 6100897 6100898 rs538 0 -
chr1 93151988 93151989 rs546 0 +
chr1 15220328 15220329 rs549 0 +
chr1 203744004 203744005 rs568 0 +
chr1 23854550 23854551 rs665 0 -
chr1 53213656 53213657 rs672 0 +
chr1 173907422 173907423 rs677 0 -
当然还有那个ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/00-All.vcf.gz 18G的文件,是VCF格式
##fileformat=VCFv4.0
##fileDate=20150218
##source=dbSNP
##dbSNP_BUILD_ID=142
##reference=GRCh38
#CHROM POS ID REF ALT QUAL FILTER INFO
1 10108 rs62651026 C T . . RS=62651026;RSPOS=10108;dbSNPBuildID=129;SSR=0;SAO=0;VP=0x050000020005000002000100;WGT=1;VC=SNV;R5;ASP
所以这个文件就是我们想要的最佳文件,提取前三列就够啦
#CHROM POS ID
1 10108 rs62651026
1 10109 rs376007522
1 10139 rs368469931
1 10144 rs144773400
1 10150 rs371194064
1 10177 rs201752861
1 10177 rs367896724
1 10180 rs201694901
1 10228 rs143255646
1 10228 rs200462216
这样就可以通过脚本用hash把我们自己找到的hash跟数据库的rs编号对应起来啦
进入网页 http://www.ncbi.nlm.nih.gov/projects/SNP/
其实就是http://www.ncbi.nlm.nih.gov/snp 这个网页
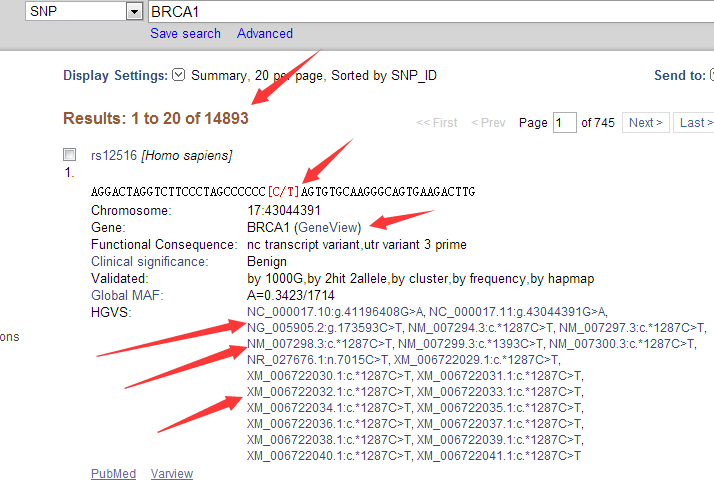
可以看到这个基因上面发表的snp非常多,共有14893个。
每个突变的各种信息都很完全,比如第一个snp位点, 它的ID是rs12516,在BRCA1基因上面。是17号染色体的43044391的碱基突变,在refseq数据库里面显示有两个NG,一个NC,还有五个NM都突变了,还有一堆XM就无所谓啦。
http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=12516
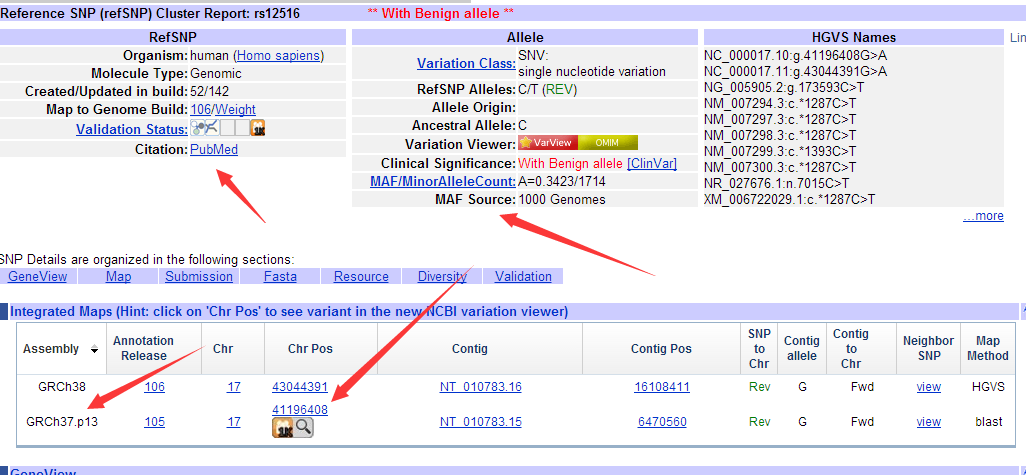
而且是有文献支持的,在1000genomes计划里面也有发表。而且是hg19和hg38里面是不同的坐标!
发表它的文献是 Associations between single nucleotide polymorphisms in double-stranded DNA repair pathway genes and familial breast cancer.
http://statgenpro.psychiatry.hku.hk/limx/kggseq/download/resources/
这个网站收集了大部分资料,我们就用它的,如果它倒闭了,大家再想办法去搜索吧。
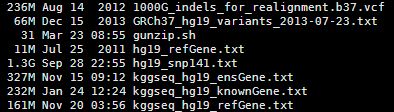
其实这些文件都是基于NCBI以及UCSC和ensembl数据库的文件用一些脚本转换而来的,都是非常简单的脚本。
首先我们看看humandb/hg19_refGene.txt 这个文件,总共2.5万多个基因的共5万多个转录本。
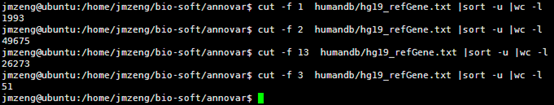
19 可能是entrez ID,但是又不像。
NM_001291929 参考基因名
chr11 染色体
-
89057521
89223909
89059923
89223852
17 89057521,89069012,89070614,89073230,89075241,89088129,89106599,89133184,89133382,89135493,89155069,89165951,89173855,89177302,89182607,89184952,89223774, 89060044,89069113,89070683,89073339,89075361,89088211,89106660,89133247,89133547,89135710,89155150,89166024,89173883,89177400,89182692,89185063,89223909,
0
NOX4 基因的英文简称,通俗名
cmpl
cmpl
2,0,0,2,2,1,0,0,0,2,2,1,0,1,0,0,0,
然后我们看看hg19_snp141.txt这个文件
1 10229 A - .
1 10229 AACCCCTAACCCTAACCCTAAACCCTA - .
1 10231 C A .
1 10231 C - .
1 10234 C T .
1 10248 A T .
1 10250 A C .
1 10250 AC - .
1 10255 A - .
1 10257 A C .
1 10259 C A .
1 10291 C T .
1 10327 T C .
1 10329 ACCCCTAACCCTAACCCTAACCCT - .
1 10330 C - .
1 10390 C - .
1 10440 C A .
1 10440 C - .
1 10469 C G .
1 10492 C T .
1 10493 C A .
1 10519 G C .
1 10583 G A 0.144169
1 10603 G A .
1 10611 C G 0.0188246
1 10617 CGCCGTTGCAAAGGCGCGCCG -
里面记录了以hg19为参考的所有的snp位点。
585
ENST00000518655 基因的ensembl ID号
chr1 + 11873 14409 14409 14409
4 基因有四个外显子
11873,12594,13402,13660, 12227,12721,13655,14409, 在基因的四个外显子的坐标
0
DDX11L1 基因的通俗英文名
none none -1,-1,-1,-1,
CTTGCCGTCAGCCTTTTCTTT·····gene的核苷酸序列
一、下载及安装软件
这个软件需要edu邮箱注册才能下载,可能是仅对科研高校开放吧。所以软件地址我就不列了。
它其实是几个perl程序,比较重要的是这个人类的数据库,snp注释必须的。
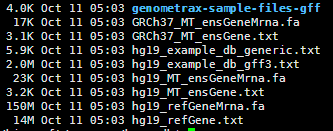
参考:http://annovar.readthedocs.org/en/latest/misc/accessory/
二,准备数据
既然是注释,那当然要有数据库啦!数据库倒是有下载地址
http://www.openbioinformatics.org/annovar/download/hg19_ALL.sites.2010_11.txt.gz
也可以用命令来下载
Perl ./annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
然后我们是对snp-calling流程跑出来的VCF文件进行注释,所以必须要有自己的VCF文件,VCF格式详解见本博客另一篇文章,或者搜索也行
http://vcftools.sourceforge.net/man_latest.html
三、运行的命令
首先把vcf格式文件,转换成空格分隔格式文件,自己写脚本也很好弄
perl convert2annovar.pl -format vcf
/home/jmzeng/raw-reads/whole-exon/snp-calling/tmp1.vcf >annovar.input
变成了空格分隔的文件
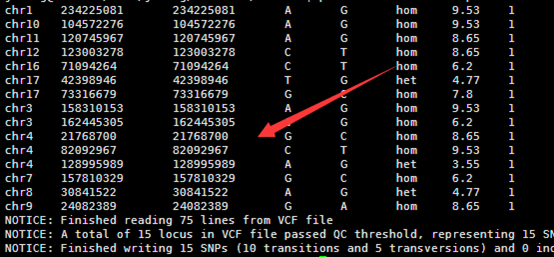
然后把转换好的数据进行注释即可
./annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/
四,输出文件解读
比对可以选择BWA或者bowtie,测序数据可以是单端也可以是双端,我这里简单讲一个,但是脚本都列出来了。而且我选择的是bowtie比对,然后单端数据。
首先进入hg19的目录,对它进行两个索引
samtools faidx hg19.fa
Bowtie2-build hg19.fa hg19
我这里随便从26G的测序数据里面选取了前1000行做了一个tmp.fa文件,进入tmp.fa这个文件的目录进行操作
Bowtie的使用方法详解见http://www.bio-info-trainee.com/?p=398
bowtie2 -x ../../../ref-database/hg19 -U tmp1.fa -S tmp1.sam
samtools view -bS tmp1.sam > tmp1.bam
samtools sort tmp1.bam tmp1.sorted
samtools index tmp1.sorted.bam
samtools mpileup -d 1000 -gSDf ../../../ref-database/hg19.fa tmp1.sorted.bam |bcftools view -cvNg - >tmp1.vcf
然后就能看到我们产生的vcf变异格式文件啦!
当然,我们可能还需要对VCF文件进行再注释!
要看懂以上流程及命令,需要掌握BWA,bowtie,samtools,bcftools,
数据格式fasta,fastq,sam,vcf,pileup
如果是bwa把参考基因组索引化,然后aln得到后缀树,然后sampe对双端数据进行比对
首先bwa index 然后选择算法,进行索引。
然后aln脚本批量处理
==> bwa_aln.sh <==
while read id
do
echo $id
bwa aln hg19.fa $id >$id.sai
done <$1
然后sampe脚本批量处理
==> bwa_sampe.sh <==
while read id
do
echo $id
bwa sampe hg19.fa $id*sai $id*single >$id.sam
done <$1
然后是samtools的脚本
==> samtools.sh <==
while read id
do
echo $id
samtools view -bS $id.sam > $id.bam
samtools sort $id.bam $id.sorted
samtools index $id.sorted.bam
done <$1
然后是bcftools的脚本
==> bcftools.sh <==
while read id
do
echo $id
samtools mpileup -d 1000 -gSDf ref.fa $id*sorted.bam |bcftools view -cvNg - >$id.vcf
done <$1
==> mpileup.sh <==
while read id
do
echo $id
samtools mpileup -d 100000 -f hg19.fa $id*sorted.bam >$id.mpileup
done <$1