R studio公司毕竟是商业化公司,在R语言推广方面做得很棒。网站什么总共有9个cheatsheet,R语言入门完全可以把这个当做笔记,写代码随时查用!
我批量下载了所有,但是想打印的时候,发现挺麻烦的,因为我不知道批量打印的方法,索性我还是半个程序猿,所以搜索了一下批量合并pdf的方法,这样就可以批量打印了,也方便传输这个文件。
其实如果在linux系统里面,一般都会自带pdf toolkit工具,里面有命令可以合并PDF文档。 Continue reading
R studio公司毕竟是商业化公司,在R语言推广方面做得很棒。网站什么总共有9个cheatsheet,R语言入门完全可以把这个当做笔记,写代码随时查用!
我批量下载了所有,但是想打印的时候,发现挺麻烦的,因为我不知道批量打印的方法,索性我还是半个程序猿,所以搜索了一下批量合并pdf的方法,这样就可以批量打印了,也方便传输这个文件。
其实如果在linux系统里面,一般都会自带pdf toolkit工具,里面有命令可以合并PDF文档。 Continue reading
这个程序是我在VirusFinder里面发现的,大家可以自行搜索它!
非常好用,建议大家写程序都可以加上这个!
print "\nChecking Java version...\n\n";my $ret = `java -version 2>&1`;print "$ret\n";if (index($ret, '1.6') == -1) {printf "Warning: The tool Trinity of the Broad Institute may require Java 1.6.\n\n";}print "\nChecking SAMtools...\n\n";$ret = `which samtools 2>&1`;if (index($ret, 'no samtools') == -1) {printf "%-30s\tOK\n\n", 'SAMtools';}else{printf "%-30s\tnot found\n\n", 'SAMtools';}my @required_modules = ("Bio::DB::Sam","Bio::DB::Sam::Constants","Bio::SeqIO","Bio::SearchIO","Carp","Config::General","Cwd","Data::Dumper","English","File::Basename","File::Copy","File::Path","File::Spec","File::Temp","FindBin","Getopt::Std","Getopt::Long","IO::Handle","List::MoreUtils","Pod::Usage","threads");print "\nChecking CPAN modules required by VirusFinder...\n\n";my $count = 0;for my $module (@required_modules){eval("use $module");if ($@) {printf "%-30s\tFailed\n", $module;$count++;}else {printf "%-30s\tOK\n", $module;}}if ($count==1){print "\n\nOne module may not be installed properly.\n\n";}elsif ($count > 1){print "\n\n$count modules may not be installed properly.\n\n";}else{print "\n\nAll CPAN modules checked!\n\n";}
其实可以手动下载local::lib, 这个perl模块,然后自己安装在指定目录,也是能解决模块的问题!
下载之后解压,进入:
$ perl Makefile.PL --bootstrap=~/.perl ##这里设置你想把模块放置的目录
$ make test && make install
$ echo 'eval $(perl -I$HOME/.perl/lib/perl5 -Mlocal::lib=$HOME/.perl)' >> ~/.bashrc
等待几个小时即可!!!
添加好环境变量之后,就可以用
perl -MCPAN -Mlocal::lib -e 'CPAN::install(LWP)'
其实你也可以直接打开 ~/.bashrc,然后写入下面的内容
PERL5LIB=$PERL5LIB:/PATH_WHERE_YOU_PUT_THE_PACKAGE/source/bin/perl_module; export PERL5LIB
可以把perl模块安装在任何地方,然后通过这种方式去把模块加载到你的perl程序!
不管别人怎么说,反正我是非常喜欢perl语言的!
也会继续学习,以前写过不少perl模块的博客,发现有点乱,正好最近看到了关于local::lib这个模块。
居然是用来解决没有root权限的用户安装,perl模块问题的!
首先说一下,如果是root用户,模块其实没有问题,直接用cpan下载器,几乎能解决所有的模块下载安装问题!
但是如果是非root用户,那么就麻烦了,很难用自动的cpan下载器,这样只能下载模块源码,然后编译,但是编译有个问题,很多模块居然是依赖于其它模块的,你的不停地下载其它依赖模块,最后才能解决,特别麻烦
但是,只需要运行下面的代码:
wget -O- http://cpanmin.us | perl - -l ~/perl5 App::cpanminus local::lib eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib` echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profile echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profile
就能拥有一个私人的cpan下载器,~/.profile可能需要更改为.bash_profile, .bashrc, etc等等,取决于你的linux系统!
然后你直接运行cpanm Module::Name,就跟root用户一样的可以下载模块啦!
或者用下面的方式在shell里面安装模块,其中ext是模块的安装目录,可以修改
perl -MTime::HiRes -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Time::HiRes;
perl -MFile::Path -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Path;
perl -MFile::Basename -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Basename;
perl -MFile::Copy -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Copy;
perl -MIO::Handle -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext IO::Handle;
perl -MYAML::XS -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML::XS;
perl -MYAML -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML;
perl -MXML::Simple -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext XML::Simple;
perl -MStorable -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Storable;
perl -MStatistics::Descriptive -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Statistics::Descriptive;
perl -MTie::IxHash -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Tie::IxHash;
perl -MAlgorithm::Combinatorics -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Algorithm::Combinatorics;
perl -MDevel::Size -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Devel::Size;
perl -MSort::Key::Radix -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key::Radix;
perl -MSort::Key -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key;
perl -MBit::Vector -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Bit::Vector;
perl -M"feature 'switch'" -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext feature;
下面是解释为什么这样可以解决问题!!!
What follows is a brief explanation of what the commands above do.
wget -O- http://cpanmin.us fetches the latest version of cpanm and prints it to STDOUT which is then piped to perl - -l ~/perl5 App::cpanminus local::lib. The first - tells perl to expect the program to come in on STDIN, this makes perl run the version of cpanm we just downloaded.perl passes the rest of the arguments to cpanm. The -l ~/perl5 argument tells cpanm where to install Perl modules, and the other two arguments are two modules to install. [App::cpanmins]1 is the package that installs cpanm. local::lib is a helper module that manages the environment variables needed to run modules in local directory.
After those modules are installed we run
eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`to set the environment variables needed to use the local modules and then
echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profileto ensure we will be able to use them the next time we log in.
echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profilewill hopefully cause man to find the man pages for your local modules.
这种类似的问题被问的特别多!
There's the way documented in perlfaq8, which is what local::lib is doing for you.
It's also a frequently asked StackOverflow question:
因为大部分生物信息学软件都是linux版本的,所以生物信息学数据分析工作者必备技能就是linux,但是大部分人只是拿他当个中转站,我以前也是,直到接触了大批量的任务,自动化流程,才明白这里面的水太深了,不过无所谓,凭我个人的观点,其实shell的进阶语法真的不必要!
我也不想去搞明白操作符两边是否加空格的区别是什么了。
如果,还有其它功能需要实现,我们可以把脚本写的更负载,纯粹的用shell,需要搜索更多的shell技巧。
[perl]
## perl nohup.pl deep_count.sh 0
## perl nohup.pl deep_count.sh 1
## perl nohup.pl deep_count.sh 2
$i=1;
open FH,$ARGV[0];
while(<FH>){
chomp;
next unless $.%3==$ARGV[1];
$cmd="nohup $_ &";
print "$cmd\n";
system($cmd);
sleep(10800) if $i%5==4;
$i++;
#exit;
}
[/perl]
如何使用自己写的私人模块
/www/modules/MyMods/Foo.pm
/www/modules/MyMods/Bar.pmAnd then where I use them:
use lib qw(/www/modules);useMyMods::Foo; useMyMods::Bar;
As reported by "perldoc -f use":
It is exactly equivalent to
BEGIN { require Module; import Module LIST; }
except that Module must be a bareword.
Putting that another way, "use" is equivalent to:
require-ing that file name, andimport-ing that package.So, instead of calling use, you can call require and import inside a BEGIN block:
BEGIN{require'../EPMS.pm';
EPMS->import();}And of course, if your module don't actually do any symbol exporting or other initialization when you call import, you can leave that line out:
BEGIN{require'../EPMS.pm';}大家看看就好,这个模块写的不怎么样,而且有高手已经写了一个pdftoolkit就是完全用这个模块实现了大部分pdf文档的操作
PDF::API2模块使用笔记
一:简单使用方法
use PDF::API2;
| # Create a blank PDF file | $pdf = PDF::API2->new(); |
| # Open an existing PDF file | $pdf = PDF::API2->open('some.pdf'); |
| # Add a blank page | $page = $pdf->page(); |
| # Retrieve an existing page | $page = $pdf->openpage($page_number); |
| # Set the page size | $page->mediabox('Letter'); |
| # Add a built-in font to the PDF | $font = $pdf->corefont('Helvetica-Bold'); |
| # Add an external TTF font to the PDF | $font = $pdf->ttfont('/path/to/font.ttf'); |
|
# Add some text to the page |
$text = $page->text();
$text->font($font, 20); $text->translate(200, 700); $text->text('Hello World!'); |
| # Save the PDF | $pdf->saveas('/path/to/new.pdf'); |
实例:
use PDF::API2;
$pdf=PDF::API2->new;
$pdf->mediabox('A4');
$ft=$pdf->cjkfont('Song');
$page = $pdf->page;
$gfx=$page->gfx;
$gfx->textlabel(50,750,$ft,20,"\x{Cool44}\x{4EA7}"); # 资产二字
$pdf->saveas('Song_Test.pdf');
二:主要对象及方法
1、pdf对象可以创造,可以打开,可以保存,可以更新,还有一堆参数可以设置
$pdf->preferences(%options)还可以设置一些浏览参数,不过本来pdf阅读器可以设置,没必要在这里花时间。
这个可以当做是个人创建pdf的保密信息,也许有一点用吧。
还可以可以设置页脚$pdf->pageLabel($index, $options
2、Page对象,可以新建,可以打开,可以保存(需要指定保存的位置)
$page = $pdf->page()
$page = $pdf->page($page_number)
$page = $pdf->openpage($page_number);
还可以更新旧的pdf,这样可以循环获取pdf页面不停的累积到一个新的pdf
$page = $pdf->import_page($source_pdf, $source_page_number, $target_page_number)
$pdf = PDF::API2->new();
$old = PDF::API2->open('our/old.pdf'); # Add page 2 from the old PDF as page 1 of the new PDF
$page = $pdf->import_page($old, 2);
$pdf->saveas('our/new.pdf');If $source_page_number is 0 or -1, it will return the last page in the document.
$count = $pdf->pages()Returns the number of pages in the document.
这样就可以写一个简单程序把我们的pdf文件合并
use PDF::API2;
my $new = PDF::API2->new;
foreach my $filename (@ARGV) { my $pdf = PDF::API2->open($filename); $new->importpage($pdf, $_) foreach 1 .. $pdf->pages;}$new->saveas('new.pdf'); $pdf->mediabox($name)
可以指定A4,A3,A5等等$pdf->mediabox($w, $h)可以指定宽度和高度$pdf->mediabox($llx, $lly, $urx, $ury)
3,还可以随意画点线面及表格,太复杂了就不看了
perl这个语言已经过时很久了,所以它的模块支持性能不是很好,暂时我只看到了对excel2003格式的表格的读取写入操作。
以下是我参考Spreadsheet::ParseExcel这个模块写的一个把excel表格转为csv的小程序,大家也可以自己搜索该模块的说明文档,这样学的更快一点!
[perl]
#!/usr/bin/perl -w
# For each tab (worksheet) in a file (workbook),
# spit out columns separated by ",",
# and rows separated by c/r.
use Spreadsheet::ParseExcel;
use strict;
use utf8;
use Encode::Locale qw($ENCODING_LOCALE_FS);
use Encode;
my $filename ="test.xls";#输入需要解析的excel文件名,必须是03版本的
my $e = new Spreadsheet::ParseExcel; #新建一个excel表格操作器
my $eBook = $e->Parse($filename); #用表格操作器来解析我们的文件
my $sheets = $eBook->{SheetCount}; #得到该文件中sheet总数
my ($eSheet, $sheetName);
foreach my $sheet (0 .. $sheets - 1) {
$eSheet = $eBook->{Worksheet}[$sheet];
$sheetName = $eSheet->{Name};
my $f1 = encode(locale_fs => $sheetName); #每次操作中文我都很纠结,还得各种转码
open FH_out ,">$f1.csv" or die "error open ";
next unless (exists ($eSheet->{MaxRow}) and (exists ($eSheet->{MaxCol})));
foreach my $row ($eSheet->{MinRow} .. $eSheet->{MaxRow}) {
foreach my $column ($eSheet->{MinCol} .. $eSheet->{MaxCol}) {
if (defined $eSheet->{Cells}[$row][$column])
{
print FH_out $eSheet->{Cells}[$row][$column]->Value . ",";
} else {
print FH_out ",";
}
}
print FH_out "\n";
}
close FH_out;
}
exit;
[/perl]
之所以要讲一下这个,是因为trinity这个软件居然需要perl的模块才能使用,所以我必须自己安装几个模块。此教程可能过期了,请直接看最新版(perl模块安装大全)
前提是要有root权限,否则只能自己下载perl模块自己解压安装了。 Continue reading
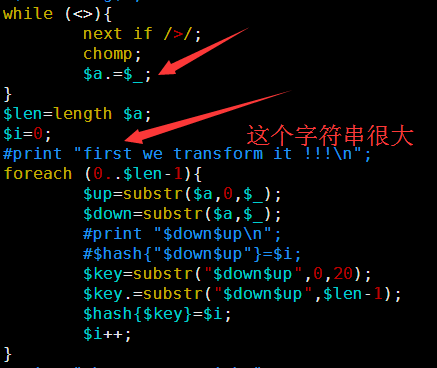
其中第一讲我提到了一个简单的索引产生方式,因为是课堂就半个小时想的,很多细节没有考虑到,对病毒那种几K大小的基因组来说是很简单的,速度也非常快,但是我测试了一下酵母,却发现好几个小时都没有结果,我只好kill掉重新改写算法,我发现之前的测序最大的问题在于没有立即substr函数的实现方式,把一个5M的字符串不停的截取首尾字符串好像是一个非常慢的方式。
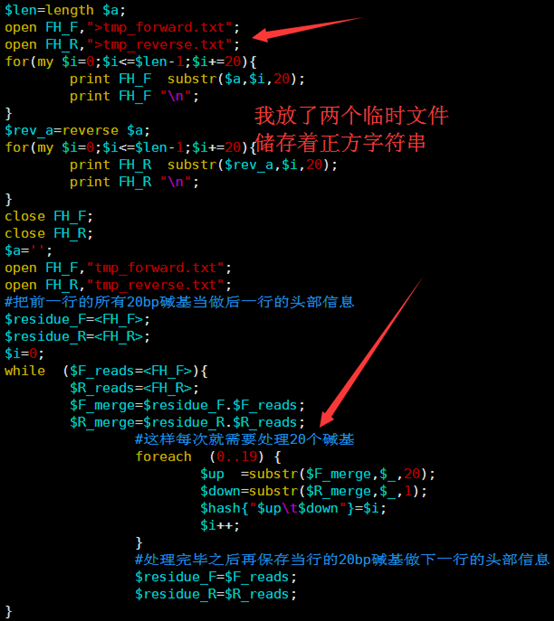
所以我优化了那个字符串的函数,虽然代码量变多了,实现过程也繁琐了一点,但是速度提升了几千倍。
time perl bwt_new_index.pl e-coli.fa >e-coli.index
测试了一下我的脚本,对酵母这样的5M的基因组,索引耗费时间是43秒
real 0m43.071s
user 0m41.277s
sys 0m1.779s
输出的index矩阵如下,我简单的截取头尾各10行给大家看,一点要看懂这个index。
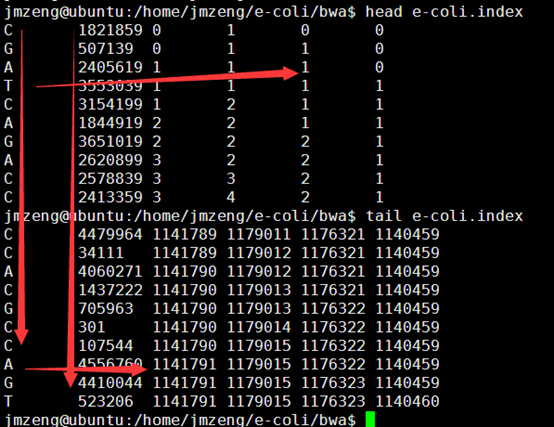
首先第一列就是我们的BWT向量,也就是BWT变换后的尾字符
第二列是之前的顺序被BWT变换后的首字符排序后的打乱的顺序。
第三,四,五,六列分别是A,C,G,T的计数,就是在当行之前累积出现的A,C,G,T的数量,是对第一列的统计。
这个索引文件将会用于下一步的查询,这里贴上我新的索引代码,查询见下一篇文章
[perl]
while (<>){
next if />/;
chomp;
$a.=$_;
}
$len=length $a;
open FH_F,">tmp_forward.txt";
open FH_R,">tmp_reverse.txt";
for(my $i=0;$i<=$len-1;$i+=20){
print FH_F substr($a,$i,20);
print FH_F "\n";
}
$rev_a=reverse $a;
for(my $i=0;$i<=$len-1;$i+=20){
print FH_R substr($rev_a,$i,20);
print FH_R "\n";
}
close FH_F;
close FH_R;
$a='';
open FH_F,"tmp_forward.txt";
open FH_R,"tmp_reverse.txt";
#把前一行的所有20bp碱基当做后一行的头部信息
$residue_F=<FH_F>;
$residue_R=<FH_R>;
$i=0;
while ($F_reads=<FH_F>){
$R_reads=<FH_R>;
$F_merge=$residue_F.$F_reads;
$R_merge=$residue_R.$R_reads;
#这样每次就需要处理20个碱基
foreach (0..19) {
$up =substr($F_merge,$_,20);
$down=substr($R_merge,$_,1);
$hash{"$up\t$down"}=$i;
$i++;
}
#处理完毕之后再保存当行的20bp碱基做下一行的头部信息
$residue_F=$F_reads;
$residue_R=$R_reads;
}
#print "then we sort it\n";
$count_a=0;
$count_c=0;
$count_g=0;
$count_t=0;
foreach (sort keys %hash){
$first=substr($_,0,1);
$len=length;
$last=substr($_,$len-1,1);
#print "$first\t$last\t$hash{$_}\n";
$count_a++ if $last eq 'A';
$count_c++ if $last eq 'C';
$count_g++ if $last eq 'G';
$count_t++ if $last eq 'T';
print "$last\t$hash{$_}\t$count_a\t$count_c\t$count_g\t$count_t\n";
}
unlink("tmp_forward.txt");
unlink("tmp_reverse.txt");
[/perl]
这里我选取酵母基因组来组装,以为它只有一条染色体,而且本身也不大!
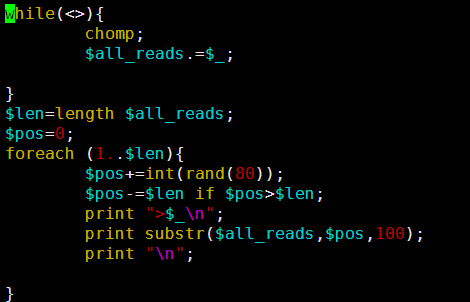
这个文件就4.5M,然后第一行就是序列名,第二列就是序列的碱基组成。共4641652个碱基。
我写一个perl程序来人为的创造一个测序文件
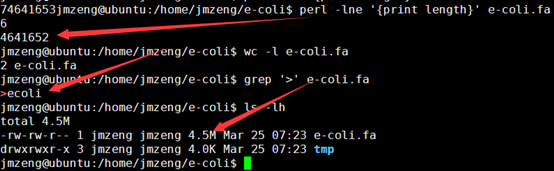
这样我们的4.5M基因组就模拟出来了486M的单端100bp的测序数据,而且是无缝连接,按照道理应该很容易就拼接的。
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer
all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
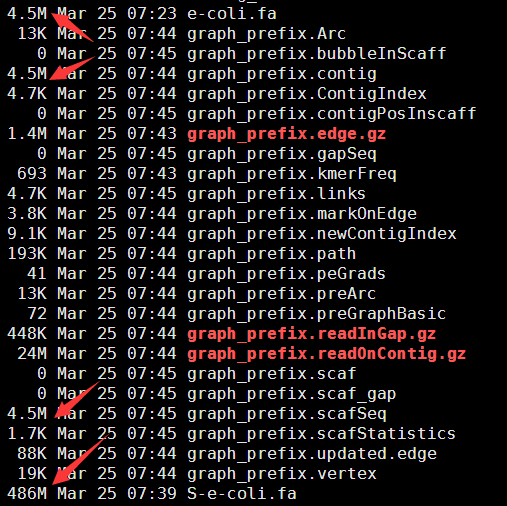
可以看到组装效果还不错哦,然后我模拟了一个测试数据,再进行组装一次,这次更好!
其实还可以模拟双端测序,应该就能达到百分百组装了。
但是由于我代码里面选取的是80在随机错开,所以我把kmer的长度设置成了81来试试看,希望这样可以把它完全组装成一条e-coli基因组。
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer
all -s config_file -K 81 -R -o graph_prefix 1>ass.log 2>ass.err
但是也没有什么实质性的提高,虽然理论上是肯定可以组装到一起!
那我再模拟一个双端测序吧,中间间隔200bp的。
需要插件和自己修改主题下面的foot.php代码。
参考 http://jingyan.baidu.com/article/ae97a646ce37c2bbfd461d01.html
步骤如下:
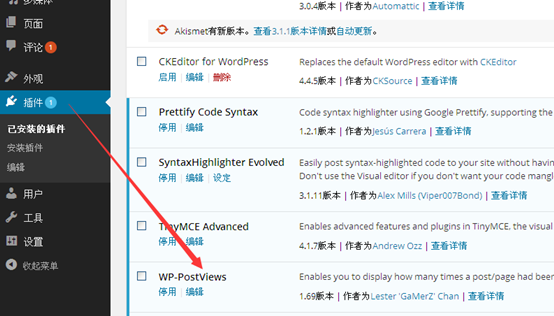
1、登陆到wp后台,鼠标移动到左侧菜单的“插件”链接上,会弹出子菜单,点击子菜单的“安装插件”链接
2、WP-PostViews插件显示wordpress文章点击浏览量
在“安装插件”链接页面的搜索框中输入“WP-PostViews”,然后回车
3、WP-PostViews插件显示wordpress文章点击浏览量
在搜索结果页面点击“WP-PostViews”插件内容区域的“现在安装”按钮
4、WP-PostViews插件显示wordpress文章点击浏览量
程序自动下载插件到服务器并解压安装,一直等到安装成功信息出现,然后在安装成功提示页面点击“启动插件”链接。
5、WP-PostViews插件显示wordpress文章点击浏览量
页面会自动跳转到“已安装插件”页面,在已安装插件列表中我们可以看到“Form Manager”插件已经处于启用状态(插件名下是“停用”链接)。
有了这个插件之后,我们的整个网页环境里面就多了一个 the_views()函数,它统计着每个文章的点击数,这样我们之前的网页就能显示点击数了。
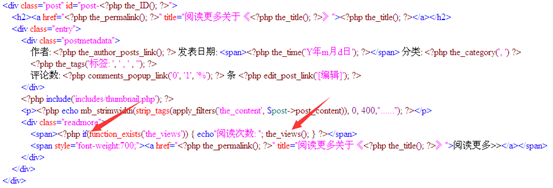
这个是我现在用的主题的php代码,把文章用span标记隔开了,而且显示着上面php代码里面的每一个内容包括日期,分类,标签,评论等等
其中thez-view()这个函数返回的不仅仅是一个访客数,但是我的文章的访客都太少了,所以我写了一个脚本帮我刷一刷流量。
[perl]
use List::MoreUtils qw(uniq);
$page='http://www.bio-info-trainee.com/?paged=';
foreach (1..5){ #我的文章比较少,就42个,所以只有5个页面
$url_page=$page.$_;
$tmp=`curl $url_page`;
#@p=$tmp=~/p=(\d+)/;
$tmp =~ s/(p=\d+)/push @p, $1/eg; #寻找p=数字这样的标签组合成新的网页地址
}
@p=uniq @p;
print "$_\n" foreach @p; #可以找到所有42个网页的地址
foreach (@p){
$new_url='http://www.bio-info-trainee.com/?'.$_;
`curl $new_url` foreach (1..100); #每个网页刷一百次
}
[/perl]
大家可以看到这个网页被刷的过程,从15到21到27直到100
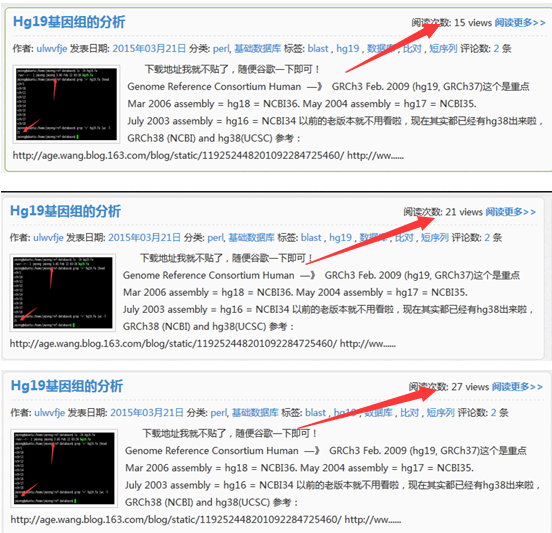
大家现在再去看我的网页,就每个文章都有一百的访问量啦!
http://www.bio-info-trainee.com/
以前的一下perl代码分享
今天去参加了开源中国的一个源创会,感觉好隆重的样子,近五百人,BAT的工程师都过来演讲了,可都是数据库相关的, 我一个的都没有听懂,但是茶歇的披萨我倒是吃了不少。
说到开源中国,我想起来了我以前在上面分享的代码,上去看了看,竟然有那么多的访问量了,让我蛮意外的,那些代码完全是我学习perl的历程的真实写照。
首先,什么是BWT,可以参考博客
http://www.cnblogs.com/xudong-bupt/p/3763814.html
他讲的非常好。
一个长度为n的串A1A2A3...An经过旋转可以得到
A1A2A3...An
A2A3...AnA1
A3...AnA1A2
...
AnA1A2A3...
n个串,每个字符串的长度都是n。
对这些字符串进行排序,这样它们之前的顺序就被打乱了,打乱的那个顺序就是index,需要输出。
首先我们测试一个简单的字符串acaacg$,总共六个字符,加上一个$符号,下次再讲$符号的意义。
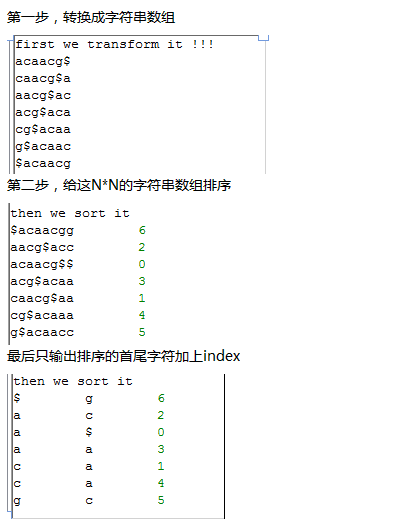
实现以上功能是比较简单的,代码如下

但是这是对于6个字符串等小片段字符串,如果是是几千万个字符的字符串,这样转换就会输出千万的平方个字符串组成的正方形数组,是很恐怖的数据量。所以在转换的同时就不能把整个千万字符储存在内存里面。
在生物学领域,是这样的,这千万个 千万个碱基的方阵,我们取每个字符串的前20个字符串就足以对它们进行排序,当然这只是近视的,我后面会讲精确排序,而且绕过内存的方法。
Perl程序如下
[perl]
while (<>){
next if />/;
chomp;
$a.=$_;
}
$a.='$';
$len=length $a;
$i=0;
print "first we transform it !!!\n";
foreach (0..$len-1){
$up=substr($a,0,$_);
$down=substr($a,$_);
#print "$down$up\n";
#$hash{"$down$up"}=$i;
$key=substr("$down$up",0,20);
$key=$key.”\t”.substr("$down$up",$len-1);
$hash{$key}=$i;
$i++;
}
print "then we sort it\n";
foreach (sort keys %hash){
$first=substr($_,0,1);
$len=length;
$last=substr($_,$len-1,1);
#print "$first\t$last\t$hash{$_}\n";
print "$_\t$hash{$_}\n";
}
[/perl]
运行的结果如下
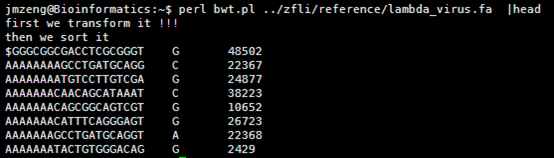
个人觉得这样排序是极好的,但是暂时还没想到如何解决不够精确的问题!!!
参考:
http://tieba.baidu.com/p/1504205984
http://www.cnblogs.com/xudong-bupt/p/3763814.html
生信常用论坛seq-answer里面所有帖子爬取
这个是爬虫专题第二集,主要讲如何分析seq-answer这个网站并爬去所有的帖子列表,及标签列表等等,前提是读者必须掌握perl,然后学习perl的LWP模块,可以考虑打印那本书读读,挺有用的!
其实爬虫是个人兴趣啦,跟这个网站没多少关系,本来一个个下载,傻瓜式的重复也能达到目的。我只是觉得这样很有技术范,哈哈,如何大家不想做傻瓜式的操作可以自己学习学习,如果不懂也可以问问我!
http://seqanswers.com/这个是主页
http://seqanswers.com/forums/forumdisplay.php?f=18 这个共570个页面需要爬取
其中f=18 代表我们要爬去的bioinformatics板块里面的内容
http://seqanswers.com/forums/forumdisplay.php?f=18&order=desc&page=1
http://seqanswers.com/forums/forumdisplay.php?f=18&order=desc&page=570
<tbody id="threadbits_forum_18">这个里面包围这很多<tr>对,
前五个<tr>对可以跳过,里面的内容不需要
生信常用论坛bio-star里面所有帖子爬取
这个是爬虫专题第一集,主要讲如何分析bio-star这个网站并爬去所有的帖子列表,及标签列表等等,前提是读者必须掌握perl,然后学习perl的LWP模块,可以考虑打印那本书读读,挺有用的!
http://seqanswers.com/ 这个是首页
http://seqanswers.com/forums/forumdisplay.php?f=18 这个共570个页面需要爬取
http://seqanswers.com/forums/forumdisplay.php?f=18&order=desc&page=1
http://seqanswers.com/forums/forumdisplay.php?f=18&order=desc&page=570
<tbody id="threadbits_forum_18">这个里面包围这很多<tr>对,
前五个<tr>对可以跳过,里面的内容不需要

仿写fastqc软件的部分功能(上)
前面我们介绍了fastqc这个软件的使用方法 http://www.bio-info-trainee.com/?p=95 ,这是一个java软件,但是有些人服务器没有配置好这个java环境,导致无法使用,这里我贴出几个perl代码,也能实现fastqc的部分功能
统一测试文件是illumina的phred33格式的fastq文件,共100000/4=25000条reads,读长都是101个碱基
程序名-fastq2quality.pl
使用命令:perl fastq2quality.pl SRR504517_1.fastq >quality.txt
功能: 把fastq格式的每条原始reads的第四行ascii码质量值,转换为Q值并输出一个矩阵,有多少条reads就有多少行,每条reads的碱基数就是列数。