首先,什么是BWT,可以参考博客
http://www.cnblogs.com/xudong-bupt/p/3763814.html
他讲的非常好。
一个长度为n的串A1A2A3...An经过旋转可以得到
A1A2A3...An
A2A3...AnA1
A3...AnA1A2
...
AnA1A2A3...
n个串,每个字符串的长度都是n。
对这些字符串进行排序,这样它们之前的顺序就被打乱了,打乱的那个顺序就是index,需要输出。
首先我们测试一个简单的字符串acaacg$,总共六个字符,加上一个$符号,下次再讲$符号的意义。
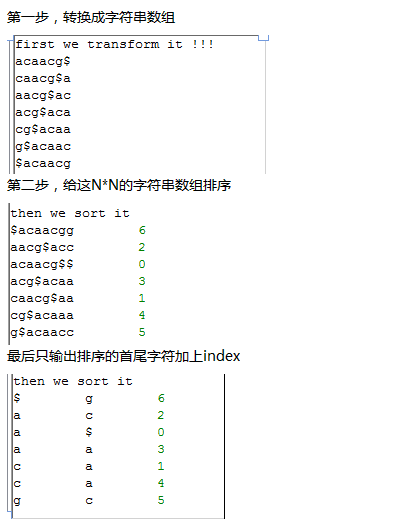
实现以上功能是比较简单的,代码如下

但是这是对于6个字符串等小片段字符串,如果是是几千万个字符的字符串,这样转换就会输出千万的平方个字符串组成的正方形数组,是很恐怖的数据量。所以在转换的同时就不能把整个千万字符储存在内存里面。
在生物学领域,是这样的,这千万个 千万个碱基的方阵,我们取每个字符串的前20个字符串就足以对它们进行排序,当然这只是近视的,我后面会讲精确排序,而且绕过内存的方法。
Perl程序如下
[perl]
while (<>){
next if />/;
chomp;
$a.=$_;
}
$a.='$';
$len=length $a;
$i=0;
print "first we transform it !!!\n";
foreach (0..$len-1){
$up=substr($a,0,$_);
$down=substr($a,$_);
#print "$down$up\n";
#$hash{"$down$up"}=$i;
$key=substr("$down$up",0,20);
$key=$key.”\t”.substr("$down$up",$len-1);
$hash{$key}=$i;
$i++;
}
print "then we sort it\n";
foreach (sort keys %hash){
$first=substr($_,0,1);
$len=length;
$last=substr($_,$len-1,1);
#print "$first\t$last\t$hash{$_}\n";
print "$_\t$hash{$_}\n";
}
[/perl]
运行的结果如下
个人觉得这样排序是极好的,但是暂时还没想到如何解决不够精确的问题!!!
参考:
http://tieba.baidu.com/p/1504205984
http://www.cnblogs.com/xudong-bupt/p/3763814.html