本来搞差异分析的工具和包就一大堆了,而且limma那个包已经非常完善了,我是不准备再讲这个的,正好有个同学问了一下这个包,我就随手测试了一下,顺便看看它跟limma有什么差异没有!手痒了就记录了测试流程!
学习一个包其实非常简单,就是找到包的官网看看说明书即可!说明书链接
本来搞差异分析的工具和包就一大堆了,而且limma那个包已经非常完善了,我是不准备再讲这个的,正好有个同学问了一下这个包,我就随手测试了一下,顺便看看它跟limma有什么差异没有!手痒了就记录了测试流程!
学习一个包其实非常简单,就是找到包的官网看看说明书即可!说明书链接
前面讲到affy处理的芯片平台是有限的,一般是hgu 95系列和133系列,[HuGene-1_1-st] Affymetrix Human Gene 1.1 ST Array这个平台虽然也是affymetrix公司的,但是affy包就无法处理 了,这时候就需要oligo包了!
oligo包是R语言的bioconductor系列包的一个,就一个功能,读取affymetix的基因表达芯片数据-CEL格式数据,处理成表达矩阵!!!
Affymetrix的探针(proble)一般是长为25碱基的寡聚核苷酸;探针总是以perfect match 和mismatch成对出现,其信号值称为PM和MM,成对的perfect match 和mismatch有一个共同的affyID。
CEL文件:信号值和定位信息。
CDF文件:探针对在芯片上的定位信息
affy包是R语言的bioconductor系列包的一个,就一个功能,读取affymetix的基因表达芯片数据-CEL格式数据,处理成表达矩阵!!!
回归的本质是建立一个模型用来预测,而逻辑回归的独特性在于,预测的结果是只能有两种,true or false
在R里面做逻辑回归也很简单,只需要构造好数据集,然后用glm函数(广义线性模型(generalized linear model))建模即可,预测用predict函数。
我这里简单讲一个例子,来自于加州大学洛杉矶分校的课程
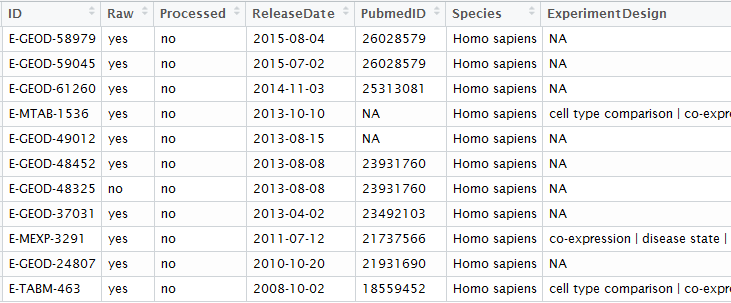
以前我写过如何使用GEOquery和GEOmetadb, 它们的确很强大,也很好用,做芯片数据pipeline的时候可以省很多力,但最近很多朋友都反应它联网有问题,经常无法下载数据!
为了解决这个问题,我仔细又研究了一下GEO数据库,其实官网本身就提供了WEB API接口,直接根据需求定制化下载数据!
我们使用GEO数据,无非就是想根据study ID号(比如:GSE1009)得到它的raw CEL文件,或者表达矩阵,或者样本分组信息!!!
如果用R包GEOquery来完成这个目的,请参考我的说明书:
其实raw CEL文件,直接自己拼接url即可
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/matrix/GSE1009_series_matrix.txt.gz
##表达矩阵,需要用在R里面read,skip掉注释信息,tab键分割
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/suppl/GSE1009_RAW.tar
##芯片原始数据,用affy包来读取
http://www.ncbi.nlm.nih.gov/geo/browse/?view=samples&series=1009&mode=csv
###样本分组信息
根据任意study ID号,非常容易就可以拼接出这些url,完全hold住GEOquery这个包的所有功能!
如果该研究涉及到的样本较多,你还可以根据下面的文件列表来有选择性的抓取样本!
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/suppl/filelist.txt
你要明白的就是浏览器的get请求而已,把下面的字符串组合成一个完整的URL即可
view=series& ## 四种,zsort=date&mode=csv& ##很重要,可以直接下载csv文件page=$i&display=5000 ##很重要查看总数:curl --silent "http://www.ncbi.nlm.nih.gov/geo/browse/" | grep "total_count"
请先看:R包精讲第一篇:如何查看你已经安装了和可以安装哪些R包?
第一种方式,当然是R自带的函数直接安装包了,这个是最简单的,而且不需要考虑各种包之间的依赖关系。
对普通的R包,直接install.packages()即可,一般下载不了都是包的名字打错了,或者是R的版本不够,如果下载了安装不了,一般是依赖包没弄好,或者你的电脑缺少一些库文件,如果实在是找不到或者下载慢,一般就用repos=来切换一些镜像。
> install.packages("ape") ##直接输入包名字即可 Installing package into ‘C:/Users/jmzeng/Documents/R/win-library/3.1’ (as ‘lib’ is unspecified) ##一般不指定lib,除非你明确知道你的lib是在哪里 trying URL 'http://mirror.bjtu.edu.cn/cran/bin/windows/contrib/3.1/ape_3.4.zip' Content type 'application/zip' length 1418322 bytes (1.4 Mb) opened URL ## 根据你选择的镜像,程序会自动拼接好下载链接url downloaded 1.4 Mb package ‘ape’ successfully unpacked and MD5 sums checked ##表明你已经安装好包啦 The downloaded binary packages are in ##程序自动下载的原始文件一般放在临时目录,会自动删除 C:\Users\jmzeng\AppData\Local\Temp\Rtmpy0OivY\downloaded_packages |
|
|
对于bioconductor的包,我们一般是
source("http://bioconductor.org/biocLite.R") ##安装BiocInstaller
#options(BioC_mirror=”http://mirrors.ustc.edu.cn/bioc/“) 如果需要切换镜像
biocLite("ggbio")或者直接BiocInstaller::biocLite('ggbio') ## 前提是你已经安装好了BiocInstaller
某些时候你还需要卸载remove.packages("BiocInstaller") 然后安装新的
第二种方式,是直接找到包的下载地址,需要进入包的主页
packageurl <- "http://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_0.9.1.tar.gz"
packageurl <- "http://cran.r-project.org/src/contrib/Archive/gridExtra/gridExtra_0.9.1.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
#packageurl <- "http://www.bioconductor.org/packages/2.11/bioc/src/contrib/ggbio_1.6.6.tar.gz"
#packageurl <- "http://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_1.0.1.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
这样安装的就不需要选择镜像了,也跨越了安装器的版本!
第三种是,先把包下载到本地,然后安装:
download.file("http://bioconductor.org/packages/release/bioc/src/contrib/BiocInstaller_1.20.1.tar.gz","BiocInstaller_1.20.1.tar.gz")
##也可以选择用浏览器下载这个包
install.packages("BiocInstaller_1.20.1.tar.gz", repos = NULL)
## 如果你用的RStudio这样的IDE,那么直接用鼠标就可以操作了
或者用choose.files()来手动交互的选择你把下载的源码BiocInstaller_1.20.1.tar.gz放到了哪里。
这种形式大部分安装都无法成功,因为R包之间的依赖性很强!
第四种是:命令行版本安装
如果是linux版本,命令行从网上自动下载包如下:
sudo su - -c \
"R -e \"install.packages('shiny', repos='https://cran.rstudio.com/')\""
如果是linux,命令行安装本地包,在shell的终端
sudo R CMD INSTALL package.tar.gz
window或者mac平台一般不推荐命令行格式,可视化那么舒心,何必自讨苦吃
这个技巧很重要,一般来说,R语言自带的install.packages函数来安装一个包时,都是用的默认的镜像!
如果你是用的Rstudio这个IDE,你的默认镜像就是: https://cran.rstudio.com/
如果你直接用的R语言,那么就是:"http://cran.us.r-project.org" 但是一般你安装的时候会提醒你选择。
既然你点进来看,肯定是有需求咯! 一般来说,R语言自带的install.packages函数来安装一个包时,都是默认安装最新版的。 但是有些R包的开发者他会引用其它的一些R包,但是它用的是人家旧版本的功能,但他自己来不及更新或者疏忽了。 而我们又不得不用他的包,这时候就不得不卸载最新版包,转而安装旧版本包。
limma真不愧是最流行的差异分析包,十多年过去了,一直是芯片数据处理的好帮手。
现在又可以支持RNA-seq数据,我赶紧试用了一下!
我下面只讲用法,大家看代码就明白了!
最近经常出现一个错误,类似于package ‘airway’ is not available (for R version 3.1.0)
就是某些包在R的仓库里面找不到,这个错误非常普遍,stackoverflow上面非常详细的解答:
在阅读这个答案的时候,我发现了一个非常有用的函数!available.packages()可以查看自己的机器可以安装哪些包!
我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
感谢读者的指正,我以前写的一个程序是错的,从算法设计上就错了!
http://www.bio-info-trainee.com/926.html
我从新设计了一个算法,经过再三检查,我可以确信它是对的,至于是否高效,就不敢保证了,也希望有更多热心的读者帮助我改正,或者跟我讨论,请直接联系我的邮箱jmzeng1314 at(防爬虫) 163.com
shiny是Rstudio公司推出的一个网页服务器,可以直接用R语言制作客户端和服务端程序来交互式的展现数据,而且有越来越丰富的扩展包可以借鉴使用,我觉得它的前途很光明,值得大家入坑!
虽然Rstudio公司吹嘘初学者无需具备html和css,js基础,但是个人认为还是多了解一下比较好,可以在w3cschool里面在线学习!
新手安装好shiny包之后里面自带了12个app例子,一边调试一边学习,很快就可以入门了。推荐一个教程,用shiny构建网页中文教程,想查看更详细的介绍和实例,请访问Shiny的官方主页。当然,你肯定需要要会R,这里有个R的FAQ教程。
如果你完全看完了上面那些,说明你已经真正入门了!
你现在可以去搜索一个shiny cheetsheet,然后背诵下来!!!!
然后你可以去shiny的github里面找到108个示例程序,一个个运行了解它们!!!
这时候你已经是高手啦!!!
接下来你应该取了解一个叫做dashboard的东西,熟练UI界面设计。
最后你再了解一些辅助包,帮助你用shiny更好的展示数据,主要是JS绘图插件
里面最出名的就是DT包了,一定要学!!!
最后你可以了解一下在rstudio上面分享五个免费的shiny网页程序,需要你搜索一些。
如果你还感兴趣的话,就可以自己整一个虚拟机Ubuntu或者centos系统,试着安装shiny的server,这个比较考验技术。
总结:你可以需要200个小时左右来完全掌握shiny
这个程序是我在VirusFinder里面发现的,大家可以自行搜索它!
非常好用,建议大家写程序都可以加上这个!
print "\nChecking Java version...\n\n";my $ret = `java -version 2>&1`;print "$ret\n";if (index($ret, '1.6') == -1) {printf "Warning: The tool Trinity of the Broad Institute may require Java 1.6.\n\n";}print "\nChecking SAMtools...\n\n";$ret = `which samtools 2>&1`;if (index($ret, 'no samtools') == -1) {printf "%-30s\tOK\n\n", 'SAMtools';}else{printf "%-30s\tnot found\n\n", 'SAMtools';}my @required_modules = ("Bio::DB::Sam","Bio::DB::Sam::Constants","Bio::SeqIO","Bio::SearchIO","Carp","Config::General","Cwd","Data::Dumper","English","File::Basename","File::Copy","File::Path","File::Spec","File::Temp","FindBin","Getopt::Std","Getopt::Long","IO::Handle","List::MoreUtils","Pod::Usage","threads");print "\nChecking CPAN modules required by VirusFinder...\n\n";my $count = 0;for my $module (@required_modules){eval("use $module");if ($@) {printf "%-30s\tFailed\n", $module;$count++;}else {printf "%-30s\tOK\n", $module;}}if ($count==1){print "\n\nOne module may not be installed properly.\n\n";}elsif ($count > 1){print "\n\n$count modules may not be installed properly.\n\n";}else{print "\n\nAll CPAN modules checked!\n\n";}
其实可以手动下载local::lib, 这个perl模块,然后自己安装在指定目录,也是能解决模块的问题!
下载之后解压,进入:
$ perl Makefile.PL --bootstrap=~/.perl ##这里设置你想把模块放置的目录
$ make test && make install
$ echo 'eval $(perl -I$HOME/.perl/lib/perl5 -Mlocal::lib=$HOME/.perl)' >> ~/.bashrc
等待几个小时即可!!!
添加好环境变量之后,就可以用
perl -MCPAN -Mlocal::lib -e 'CPAN::install(LWP)'
其实你也可以直接打开 ~/.bashrc,然后写入下面的内容
PERL5LIB=$PERL5LIB:/PATH_WHERE_YOU_PUT_THE_PACKAGE/source/bin/perl_module; export PERL5LIB
可以把perl模块安装在任何地方,然后通过这种方式去把模块加载到你的perl程序!
不管别人怎么说,反正我是非常喜欢perl语言的!
也会继续学习,以前写过不少perl模块的博客,发现有点乱,正好最近看到了关于local::lib这个模块。
居然是用来解决没有root权限的用户安装,perl模块问题的!
首先说一下,如果是root用户,模块其实没有问题,直接用cpan下载器,几乎能解决所有的模块下载安装问题!
但是如果是非root用户,那么就麻烦了,很难用自动的cpan下载器,这样只能下载模块源码,然后编译,但是编译有个问题,很多模块居然是依赖于其它模块的,你的不停地下载其它依赖模块,最后才能解决,特别麻烦
但是,只需要运行下面的代码:
wget -O- http://cpanmin.us | perl - -l ~/perl5 App::cpanminus local::lib eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib` echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profile echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profile
就能拥有一个私人的cpan下载器,~/.profile可能需要更改为.bash_profile, .bashrc, etc等等,取决于你的linux系统!
然后你直接运行cpanm Module::Name,就跟root用户一样的可以下载模块啦!
或者用下面的方式在shell里面安装模块,其中ext是模块的安装目录,可以修改
perl -MTime::HiRes -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Time::HiRes;
perl -MFile::Path -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Path;
perl -MFile::Basename -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Basename;
perl -MFile::Copy -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Copy;
perl -MIO::Handle -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext IO::Handle;
perl -MYAML::XS -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML::XS;
perl -MYAML -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML;
perl -MXML::Simple -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext XML::Simple;
perl -MStorable -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Storable;
perl -MStatistics::Descriptive -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Statistics::Descriptive;
perl -MTie::IxHash -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Tie::IxHash;
perl -MAlgorithm::Combinatorics -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Algorithm::Combinatorics;
perl -MDevel::Size -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Devel::Size;
perl -MSort::Key::Radix -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key::Radix;
perl -MSort::Key -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key;
perl -MBit::Vector -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Bit::Vector;
perl -M"feature 'switch'" -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext feature;
下面是解释为什么这样可以解决问题!!!
What follows is a brief explanation of what the commands above do.
wget -O- http://cpanmin.us fetches the latest version of cpanm and prints it to STDOUT which is then piped to perl - -l ~/perl5 App::cpanminus local::lib. The first - tells perl to expect the program to come in on STDIN, this makes perl run the version of cpanm we just downloaded.perl passes the rest of the arguments to cpanm. The -l ~/perl5 argument tells cpanm where to install Perl modules, and the other two arguments are two modules to install. [App::cpanmins]1 is the package that installs cpanm. local::lib is a helper module that manages the environment variables needed to run modules in local directory.
After those modules are installed we run
eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`to set the environment variables needed to use the local modules and then
echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profileto ensure we will be able to use them the next time we log in.
echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profilewill hopefully cause man to find the man pages for your local modules.
这种类似的问题被问的特别多!
There's the way documented in perlfaq8, which is what local::lib is doing for you.
It's also a frequently asked StackOverflow question:
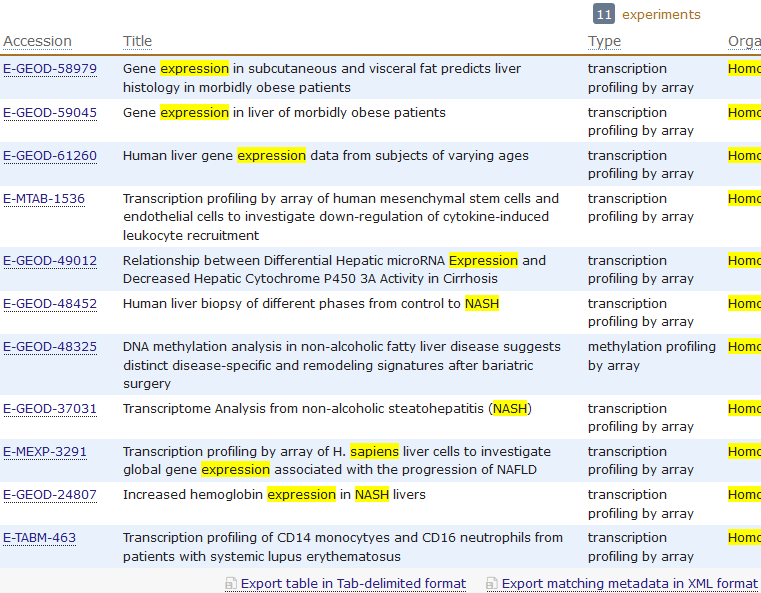
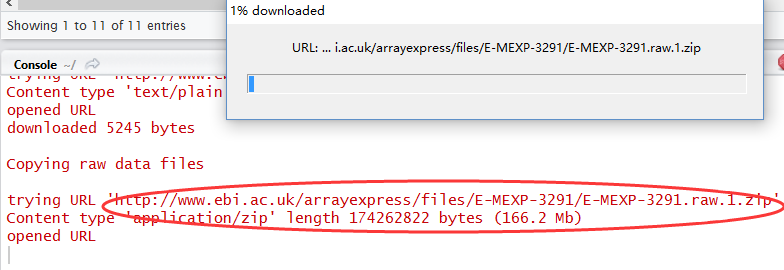
rawset = ArrayExpress("E-MEXP-3291")
现有的基因芯片种类不要太多了!
gpl organism bioc_package1 GPL32 Mus musculus mgu74a2 GPL33 Mus musculus mgu74b3 GPL34 Mus musculus mgu74c6 GPL74 Homo sapiens hcg1107 GPL75 Mus musculus mu11ksuba8 GPL76 Mus musculus mu11ksubb9 GPL77 Mus musculus mu19ksuba10 GPL78 Mus musculus mu19ksubb11 GPL79 Mus musculus mu19ksubc12 GPL80 Homo sapiens hu680013 GPL81 Mus musculus mgu74av214 GPL82 Mus musculus mgu74bv215 GPL83 Mus musculus mgu74cv216 GPL85 Rattus norvegicus rgu34a17 GPL86 Rattus norvegicus rgu34b18 GPL87 Rattus norvegicus rgu34c19 GPL88 Rattus norvegicus rnu3420 GPL89 Rattus norvegicus rtu3422 GPL91 Homo sapiens hgu95av223 GPL92 Homo sapiens hgu95b24 GPL93 Homo sapiens hgu95c25 GPL94 Homo sapiens hgu95d26 GPL95 Homo sapiens hgu95e27 GPL96 Homo sapiens hgu133a28 GPL97 Homo sapiens hgu133b29 GPL98 Homo sapiens hu35ksuba30 GPL99 Homo sapiens hu35ksubb31 GPL100 Homo sapiens hu35ksubc32 GPL101 Homo sapiens hu35ksubd36 GPL201 Homo sapiens hgfocus37 GPL339 Mus musculus moe430a38 GPL340 Mus musculus mouse430239 GPL341 Rattus norvegicus rae230a40 GPL342 Rattus norvegicus rae230b41 GPL570 Homo sapiens hgu133plus242 GPL571 Homo sapiens hgu133a243 GPL886 Homo sapiens hgug4111a44 GPL887 Homo sapiens hgug4110b45 GPL1261 Mus musculus mouse430a249 GPL1352 Homo sapiens u133x3p50 GPL1355 Rattus norvegicus rat230251 GPL1708 Homo sapiens hgug4112a54 GPL2891 Homo sapiens h20kcod55 GPL2898 Rattus norvegicus adme16cod60 GPL3921 Homo sapiens hthgu133a63 GPL4191 Homo sapiens h10kcod64 GPL5689 Homo sapiens hgug4100a65 GPL6097 Homo sapiens illuminaHumanv166 GPL6102 Homo sapiens illuminaHumanv267 GPL6244 Homo sapiens hugene10sttranscriptcluster68 GPL6947 Homo sapiens illuminaHumanv369 GPL8300 Homo sapiens hgu95av270 GPL8490 Homo sapiens IlluminaHumanMethylation27k71 GPL10558 Homo sapiens illuminaHumanv472 GPL11532 Homo sapiens hugene11sttranscriptcluster73 GPL13497 Homo sapiens HsAgilentDesign02665274 GPL13534 Homo sapiens IlluminaHumanMethylation450k75 GPL13667 Homo sapiens hgu21976 GPL15380 Homo sapiens GGHumanMethCancerPanelv177 GPL15396 Homo sapiens hthgu133b78 GPL17897 Homo sapiens hthgu133a
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)### first download all of the annotation packages from bioconductorfor (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) == FALSE) {BiocInstaller::biocLite(platformDB)#source("http://bioconductor.org/biocLite.R");#biocLite(platformDB )}}
下载完了所有的包, 就可以进行批量导出芯片探针与gene的对应关系!
for (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform)#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) != FALSE) {library(platformDB,character.only = T)#tmp=paste('head(mappedkeys(',platform,'ENTREZID))',sep='')#eval(parse(text = tmp))###重点在这里,把字符串当做命令运行all_probe=eval(parse(text = paste('mappedkeys(',platform,'ENTREZID)',sep='')))EGID <- as.numeric(lookUp(all_probe, platformDB, "ENTREZID"))##自己把内容写出来即可}}