文章:CARM1 Methylates Chromatin Remodeling Factor BAF155 to Enhance Tumor Progression and Metastasis
我很早以前想自学CHIP-seq的时候就关注过这篇文章,那时候懂得还不多,甚至都没有仔细看这篇文章就随便下载了数据进行分析,也只是跑一些软件而已,这次仔细阅读这篇文章才发现里面的门道很多,尤其是CHIP-seq的实验基础,以及表观遗传学的生物学基础知识,我有时间一定要把这篇文章翻译一下。学习这篇文章前一定要温习一些生物学知识,见我上一篇博客
作者首先实验证明了用small haripin RNA来knockout CARM1 只能达到90%的敲除效果,有趣的是,对CARM1的功能影响非常小,说明只需要极地量的CARM1就可以很好的发挥作用,所以作者设计了100%敲除CARM1的实验材料,通过zinc finger nuclease这种基因组编辑技术( 缩写成ZFN技术)。
这样就能比较CARM1有无的机体种各种蛋白被催化状态了,其中SWI/SNF(BAF) chromatin remodeling complex 染色质重构复合物的一个亚基 BAF155,非常明显的只有在CARM1这个基因完好无损的细胞系里面才能被正常的甲基化。作者证明了BAF155是CARM1这个基因非常好(拉丁语 bona fide)的一个底物, 而且通过巧妙的实验设计,证明了BAF155这个蛋白的第1064位氨基酸(R) 是 CARM1的作用位点。
因为早就有各种文献说明了SWI/SNF(BAF) chromatin remodeling complex 染色质重构复合物在癌症的重要作用, 所以作者也很自然的想探究BAF155在癌症的功能详情。这里作者选择的是CHIP-seq技术,因为BAF155是转录因子的一种。(转录因子(transcription factor)是一群能与基因5`端上游特定序列专一性结合,从而保证目的基因以特定的强度在特定的时间与空间表达的蛋白质分子。)CHIP-seq技术最适合来探究BAF155这样转录因子的功能了,所以作者构造了一种细胞系(MCF7),它的BAF155这个蛋白的第1064位氨基酸(R) 突变了,这样就无法被CARM1这个基因催化而甲基化,然后比较突变的细胞系和野生型细胞系的BAF155的CHIP-seq结果,这样就可以研究BAF155这个转录因子,是否必须要被CARM1这个基因催化而甲基化后才能行使生物学功能。
作者用me-BAF155特异性抗体+western bloting 证明了正常的野生型MCF7细胞系里面有~74%的BAF155是被甲基化的!
有一个细胞系SKOV3,可以正常表达除了BAF155之外的其余14种SWI/SNF(BAF) chromatin remodeling complex 染色质重构复合物,而不管是把突变的细胞系和野生型细胞系的BAF155混在里面都可以促进染色质重构复合物的组装,所以甲基化与否并不影响这个染色质重构复合物的组装,我们重点应该研究的是甲基化会影响BAF155在基因组其它地方结合。
结果是,突变的细胞系和野生型细胞系种BAF155在基因组结合位置(peaks)还是有较大的overlap的,重点是看它们的peaks在各种基因组区域(基因上下游,5,3端UTR,启动子,内含子,外显子,基因间区域,microRNA区域)分布情况的差别,还有它们举例转录起始位点的距离的分布区别,还有它们注释到的基因区别,已经基因富集到什么通路,等等这样的分析。
虽然作者在人的细胞系(MCF7)上面做CHIP-seq,但是在老鼠细胞系(MDA-MB-231)做了mRNA芯片数据分析,BAF155这个蛋白的第1064位氨基酸(R) 突变细胞系,和野生型细胞系,用的是Affymetrix HG U133 Plus 2.0这个常用平台
which was hybridized to Affymetrix HG U133 Plus 2.0 microarrays containing 54,675 probesets for >47,000 transcripts and variants, including 38,500 human genes.
To identify genes differentially expressed between MDA-MB-231-BAF155WT and MDA-MB-231-BAF155R1064K
我简单摘抄作者的对CHIP-seq数据的生物信息学分析结果
## All samples were mapped from fastq files using BOWTIE [-m 1 -- best] to mm9 [UCSCmouse genome build 9]
## Sequences were mapped to the human genome (hg19) using BOWTIE (--best –m 1) to yield unique alignments
## Peaks were called by using HOMER [http://biowhat.ucsd.edu/homer/] and QuEST [http://mendel.stanford.edu/sidowlab/downloads/quest/].
QuEST 2.4 (Valouev et al., 2008) was run using the recommend settings for transcription factor (TF) like binding with the following exceptions:
kde_bandwith=30, region_size=600, ChIP threshold=35, enrichment fold=3, rescue fold=3.
HOMER (Heinz et al., 2010) analysis was run using the default settings for peak finding.
False Discovery Rate (FDR) cut off was 0.001 (0.1%) for all peaks.
The tag density for each factor was normalized to 1x107 tags and displayed using the UCSC genome browser.
Motif analysis (de novo and known), was performed using the HOMER software and Genomatix.
Peak overlaps were processed with HOMER and Galaxy (Giardine et al., 2005).
Peak comparisons between replicates were processed with EdgeR statistical package in R
也就是我们接下来需要学习的流程化分析步骤,下面我给一个主要流程的截图,但是主要还是要看实验是如何设计的,也有一个文章发表关于CHIP-seq的流程的:http://biow.sb-roscoff.fr/ecole_bioinfo/protected/jacques.van-helden/ThomasChollier_NatProtoc_2012_peak-motifs.pdf
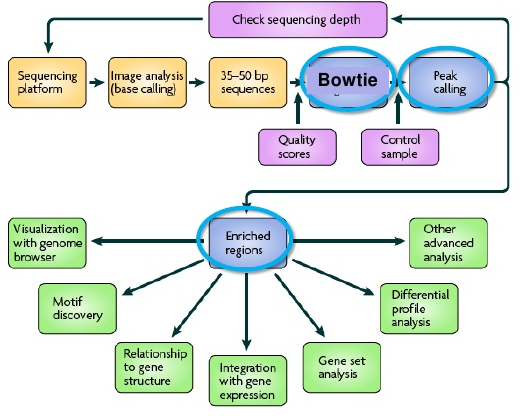
同时我还推荐大家看几篇相关文献:
Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. http://www.nature.com/nature/journal/v448/n7153/pdf/nature06008.pdf
Mapping and analysis of chromatin state dynamics in nine human cell types(GSE26386): http://www.nature.com/nature/journal/v473/n7345/full/nature09906.html
Promiscuous RNA binding by Polycomb Repressive Complex 2 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3823624/pdf/nihms517229.pdf