在使用Bowtie比对于完Chip-Seq的结果后,就需要用到MACS或者ERANGE来找出峰所在的位置了。但是由于ERANGE的设置比较复杂,所以最为流行的还是MACS。
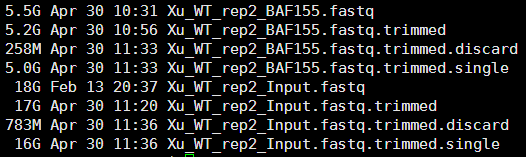
一.首先安装MACS软件
MACS有两个版本,分别是MACS14和MACS2。MACS2在很多方面都对MACS14做了重大改进,但目前还在测试阶段。我们依然以MACS14为例进行说明。
MACS软件的下载地址在wget https://codeload.github.com/taoliu/MACS/zip/master
这是一个python软件,有152M,已经算是很大了!所以需要按照安装python的方法来安装它!但是,好像这个是最新版的,我们还是用1.4版本吧
wget http://github.com/downloads/taoliu/MACS/MACS-1.4.2-1.tar.gz
其实它的readme已经把这个软件的各种安装使用方法讲的很清楚了。
https://github.com/taoliu/MACS/blob/master/README.rst
MACS软件的具体原理,大家去看文献,或者参考这篇文章
http://www.plob.org/2014/05/08/7227.html
很简单的一个python命令即可安装该软件python setup.py install --user
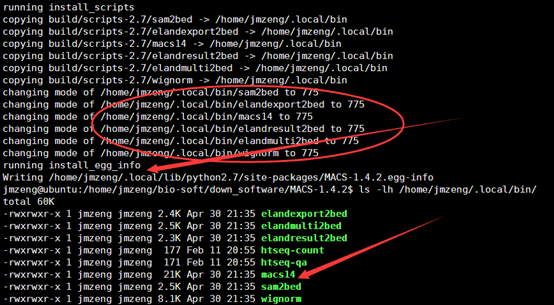
二.然后准备该软件所需要的数据
是我们在前两篇文章中提到的数据
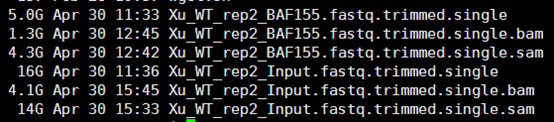
三.接着运行MACS的命令
/home/jmzeng/.local/bin/macs14 -t Xu_WT_rep2_BAF155.fastq.trimmed.single.bam \
> -c Xu_WT_rep2_Input.fastq.trimmed.single.bam \
> -f BAM -g hs --bw 300 -w -S -n Xu_WT_rep2
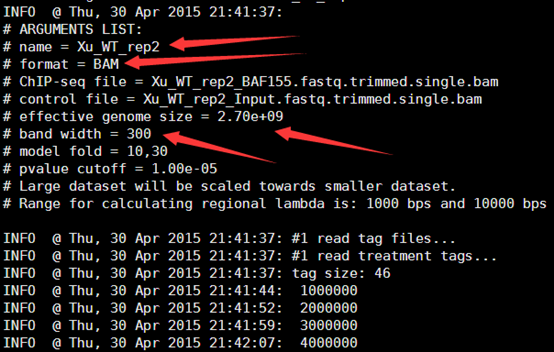
四.最后解读一下结果
56K Apr 30 21:54 Xu_WT_rep2_model.r
5.5K Apr 30 22:21 Xu_WT_rep2_negative_peaks.xls
783K Apr 30 22:21 Xu_WT_rep2_peaks.bed
865K Apr 30 22:21 Xu_WT_rep2_peaks.xls
766K Apr 30 22:21 Xu_WT_rep2_summits.bed
唉,反正这也不是我的课题,懒得解释这些结果啦,等后来有机会再慢慢玩吧
参考 http://www.plob.org/2014/05/08/7227.html
附录:我们现在来了解如何设置参数。
参考自 http://www.plob.org/2014/01/26/7118.html
-t TFILE, –treatment=TFILE 输入文件名
-c CFILE, –control=CFILE 输入阴对文件名
-n NAME, –name=NAME 输入出文件名前缀
-f FORMAT, –format=FORMAT 输入文件格式,默认值为AUTO,可选的值为”BEG”,”ELAND”,”ELANDMULTI”,”ELANDMULTIPER”,”ELANDEXPORT”,”SAM”,”BAM”,”BOWTIE”等。
-g GSIZE, –gsize=GSIZE 比对模板大小。格式可以是:1.0e+9,或者1000000000,也可以缩写:’hs’ for 人类 (2.7e9), ‘mm’ for 大鼠(1.87e9), ‘ce’ for 线虫 (9e7) and ‘dm’ for 果蝇 (1.2e8), 默认值:hs
-s TSIZE, –tsize=TSIZE 设置为短序列的长度,默认值为25
-p PVALE, –pvalue=PVALUE 非峰可能性截取值,默认值为1e-5,这个值不能大太,超过0.9的话,可能无法输出正确的结果
-m MFOLD, –mfold=MFOLD 峰值高度相对于本底的比值,默认值为10,30。也就是说,最低值不能少于10,但比值超过30也不认为它是正常的一个峰。一般而言,低值设置为10是一个很好的区分点。如果这个值还是无法得到满意的结果,那么可以设置得更低,但最好还是使用–nomodel参数,使–nomodel设置为True,然后再传递–shiftsize及–bw参数给MACS。–shiftsize默认值为100,而–bw的默认值为300。
–diag 生成完整报表,会包括是否为真峰的可能性,但会严重拖累运算速度。