通过前面的分析,我们已经量化了ET1刺激前后的细胞的miRNA和mRNA表达水平,也通过成熟的统计学分析分别得到了差异miRNA和mRNA,这时候我们就需要换一个参考文献了,因为前面提到的那篇文章分析的不够细致,我这里选择了浙江大学的一篇TCGA数据挖掘分析文章Identifying miRNA/mRNA negative regulation pairs in colorectal cancer,里面首先就是查找miRNA-mRNA基因对,因为miRNA主要还是负向调控mRNA表达,所以根据我们得到的两个表达矩阵做相关性分析,很容易得到符合统计学意义的miRNA-mRNA基因对,具体分析内容如下:
把得到的差异miRNA的表达量画一个热图,看看它是否能显著的分类
用miRWalk2.0等数据库或者根据来获取这些差异miRNA的validated target genes
然后看看这些pairs of miRNA- target genes的表达量相关系数,选取显著正相关或者负相关的pairs
这些被选取的pairs of miRNA- target genes拿去做富集分析
最后这些pairs of miRNA- target genes做PPI网络分析
首先我们看第一个热图的实现:
resOrdered=na.omit(resOrdered)
DEmiRNA=resOrdered[abs(resOrdered$log2FoldChange)>log2(1.5) & resOrdered$padj <0.01 ,]
write.csv(resOrdered,"deseq2.results.csv",quote = F)
DEmiRNAexprSet=exprSet[rownames(DEmiRNA),]
write.csv(DEmiRNAexprSet,'DEmiRNAexprSet.csv')
DEmiRNAexprSet=read.csv('DEmiRNAexprSet.csv',stringsAsFactors = F)
exprSet=as.matrix(DEmiRNAexprSet[,2:7])
rownames(exprSet)=rownames(DEmiRNAexprSet)
heatmap(exprSet)
gplots::heatmap.2(exprSet)
library(pheatmap)
## http://biit.cs.ut.ee/clustvis/
因为我前面保存的表达量就基于counts的,所以画热图还需要进行normalization,我这里懒得弄了,就用了一个网页版工具,自动出热图http://biit.cs.ut.ee/clustvis/
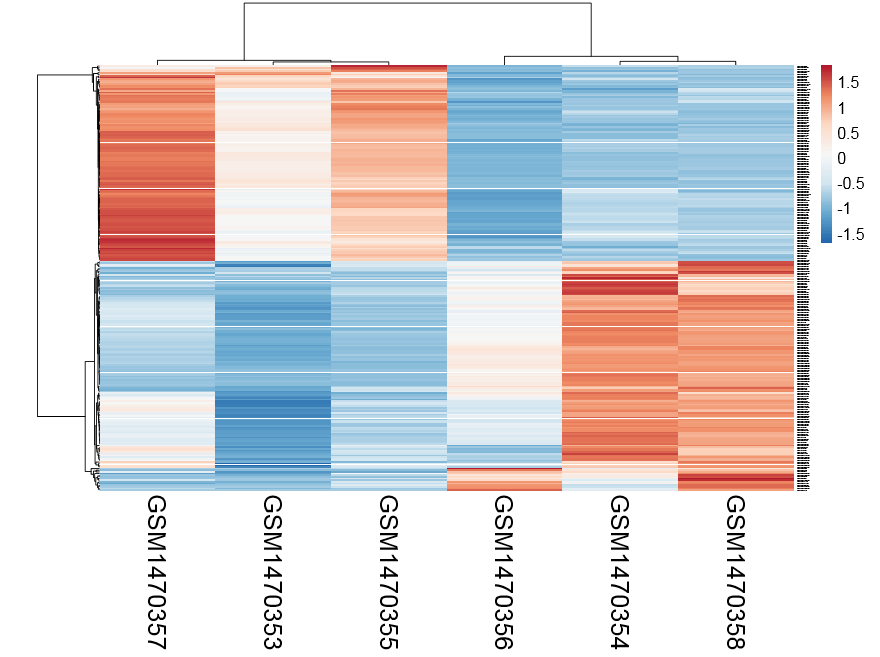
感觉还不错,可以很清楚的看到ET1刺激前后细胞中miRNA表达量变化
然后就是检验我们选取的感兴趣的有显著差异的miRNA的target genes,这时候有两种方法,一个是先由数据库得到已经被检验的miRNA的target genes,另一种是根据miRNA和mRNA表达量的相关性来预测。
用数据库来查找MiRNA的作用基因,非常多的工具,比较常用的有TargetScan/miRTarBase
### http://nar.oxfordjournals.org/content/early/2015/11/19/nar.gkv1258.full
### http://mirtarbase.mbc.nctu.edu.tw/
### http://mirtarbase.mbc.nctu.edu.tw/cache/download/6.1/hsa_MTI.xlsx
### http://www.targetscan.org/vert_71/ (version 7.1 (June 2016))
我还看到过一个整合工具: miRecords (DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid and TargetScan/TargertScanS)里面提到了查找MiRNA的作用基因这一过程,高假阳性,至少被5种工具支持,才算是真的
还有很多类似的工具,miRWalk2,psRNATarget网页版工具,最后值得一提的是中山大学的: starBase Pan-Cancer Analysis Platform is designed for deciphering Pan-Cancer Networks of lncRNAs, miRNAs, ceRNAs and RNA-binding proteins (RBPs) by mining clinical and expression profiles of 14 cancer types (>6000 samples) from The Cancer Genome Atlas (TCGA) Data Portal (all data available without limitations).虽然我没有仔细的用,但是看介绍好牛的样子,还有一个R包:miRLAB我玩了一会,它是先通过算所有配对的miRNA- genes的表达量相关系数,选取显著正相关或者负相关的pairs,然后反过来通过已知数据库来验证。
后面我就不讲了,主要看你得到miRNA的时候其它生物学数据是否充分,如果是癌症病人,有生存相关数据,可以做生存分析,如果你同时测了甲基化数据,可以做甲基化相关分析~~~~~~~~~
如果只是单纯的miRNA测序数据,可以回过头去研究一下de novo的miRNA预测的步骤,也是研究重点