这也是对TCGA数据的深度挖掘,从而提出的一个统计学概念。文章研究了30种癌症,发现21种不同的mutation signature。如果理解了,就会发现这个其实蛮简单的,他们并不重新测序,只是拿已经有了的TCGA数据进行分析,而且居然是发表在nature上面!
研究了4,938,362 mutations from 7,042 cancers样本,突变频谱的概念只是针对于somatic 的mutation。一般是对癌症病人的肿瘤组织和癌旁组织配对测序,过滤得到的somatic mutation,一般一个样本也就几百个somatic 的mutation。
paper链接是:http://www.nature.com/nature/journal/v500/n7463/full/nature12477.html
从2013年提出到现在,已经有30种mutation siganures,在cosmic数据库有详细记录,更新见:http://cancer.sanger.ac.uk/cosmic/signatures
它的概念就是:根据突变上下文分成96类,然后每类突变的频率不一样画一个条形图,可视化展现。
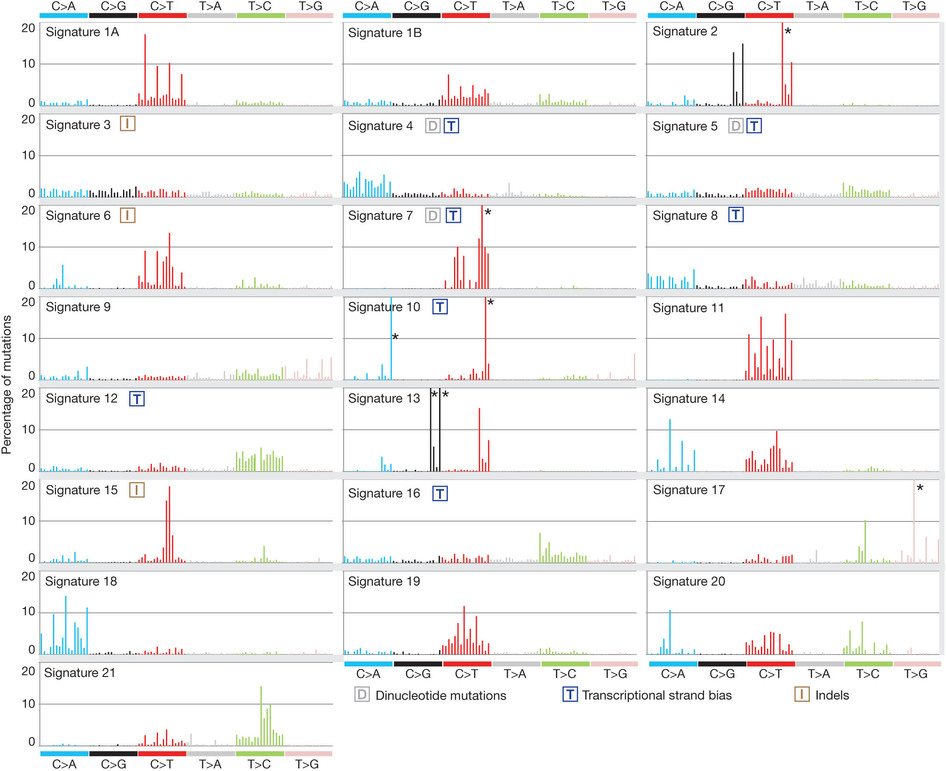
Each signature is displayed according to the 96 substitution classification defined by the substitution class and sequence context immediately 3′ and 5′ to the mutated base. The probability bars for the six types of substitutions are displayed in different colours.
仔细看paper,还是蛮好理解的,自己写一个脚本就可以做这个分析了,前提是下载各个癌症的somatic mutation文件,一般是maf格式的,很多途径下载。
In principle, all classes of mutation (such as substitutions, indels, rearrangements) and any accessory mutation characteristic, for example, the sequence context of the mutation or the transcriptional strand on which it occurs, can be incorporated into the set of features by which a mutational signature is defined. In the first instance, we extracted mutational signatures using base substitutions and additionally included information on the sequence context of each mutation. Because there are six classes of base substitution—C>A, C>G, C>T, T>A, T>C, T>G (all substitutions are referred to by the pyrimidine of the mutated Watson–Crick base pair)—and as we incorporated information on the bases immediately 5′ and 3′ to each mutated base, there are 96 possible mutations in this classification. This 96 substitution classification is particularly useful for distinguishing mutational signatures that cause the same substitutions but in different sequence contexts.
很多癌症都发现了不止一种mutation signature,甚至高达6种,说明癌症之间差异还是蛮大的!
In most cancer classes at least two mutational signatures were observed, with a maximum of six in cancers of the liver, uterus and stomach. Although these differences may, in part, be attributable to differences in the power to extract signatures, it seems likely that some cancers have a more complex repertoire of mutational processes than others.
Most individual cancer genomes exhibit more than one mutational signature and many different combinations of signatures were observed
但是,我最后也没能绝对的界限是什么,因为总不能用肉眼来看每个突变频谱不一样吧?
The set of signatures will be updated in the future. This will include incorporating additional mutation types (e.g., indels, structural rearrangements, and localized hypermutation such as kataegis) and cancer samples. With more cancer genome sequences and the additional statistical power this will bring, new signatures may be found, the profiles of current signatures may be further refined, signatures may split into component signatures and signatures may be found in cancer types in which they are currently not detected.
分类会持续不断更新,随着更多的cancer type和样本加入,新的signature会被发现,现有的signature也可能会被重新定义,或者被分割成多个小的signature