how to use GSEA?
这个有点类似于pathway(GO,KEGG等)的富集分析,区别在于gene set(矫正好的基于文献的数据库)的概念更广泛一点,包括了
how to download GSEA ?
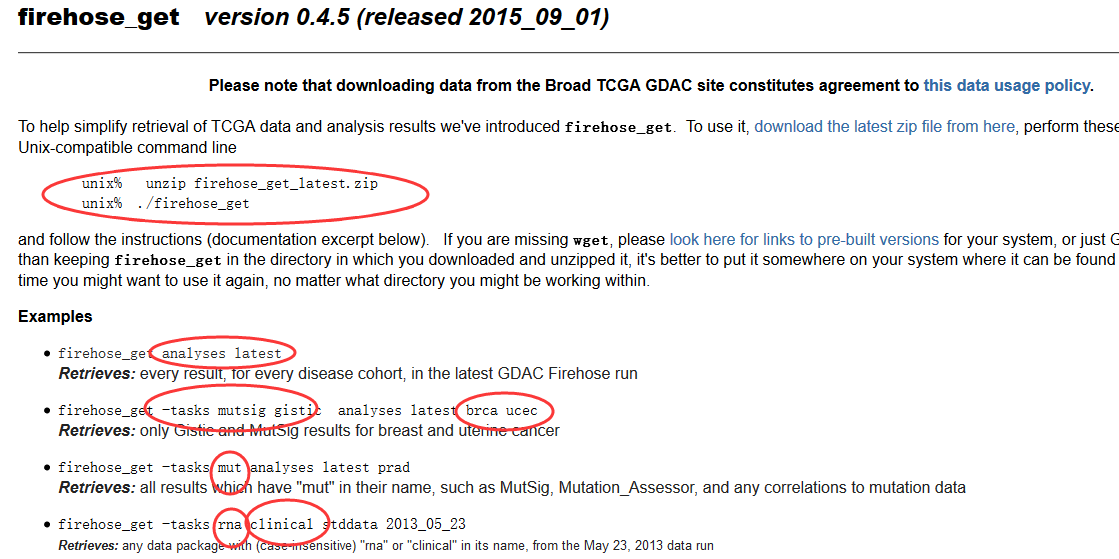
软件下载地址:http://software.broadinstitute.org/gsea/downloads.jsp
需要自己安装好java环境!
what's the input for the GSEA?
说明书上写的输入数据是:GSEA supported data files are simply tab delimited ASCII text files, which have special file extensions that identify them. For example, expression data usually has the extension *.gct, phenotypes *.cls, gene sets *.gmt, and chip annotations *.chip. Click the More on file formats help button to view detailed descriptions of all the data file formats.
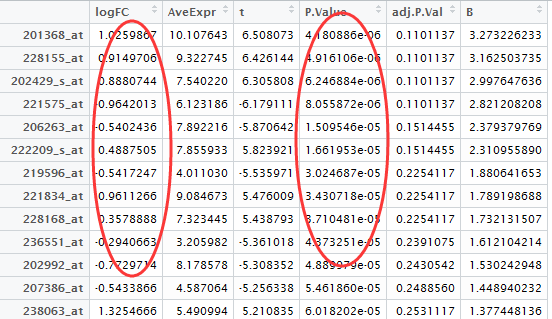
实际上没那么复杂,一个表达矩阵即可!然后做一个分组说明的cls文件即可。
主要是自己看说明书,做出要求的数据格式:http://www.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
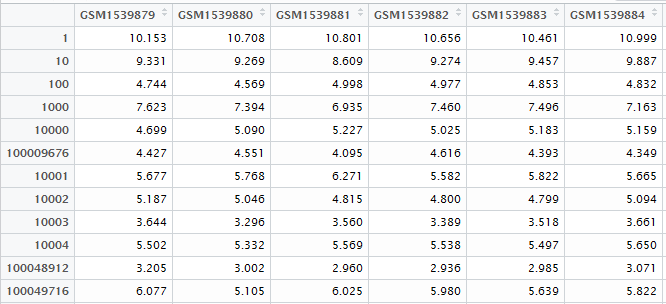
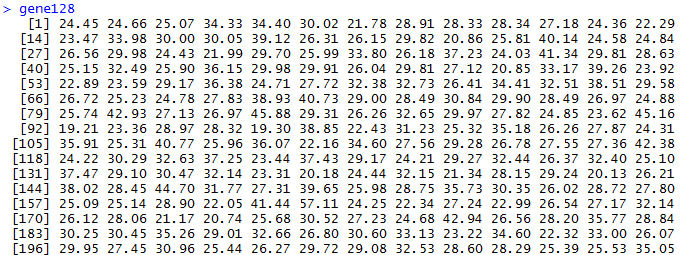
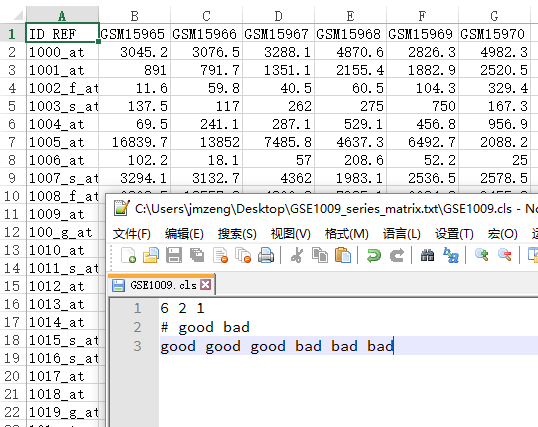
表达矩阵我这里下载GSE1009数据集做测试吧!
cls的样本说明文件,就随便搞一搞吧,下面这个是例子:
6 2 1
# good bad
good good good bad bad bad
文件如下,六个样本,根据探针来的表达数据,分组前后各三个一组。
现在开始运行GSEA!
start to run the GSEA !
首先载入数据
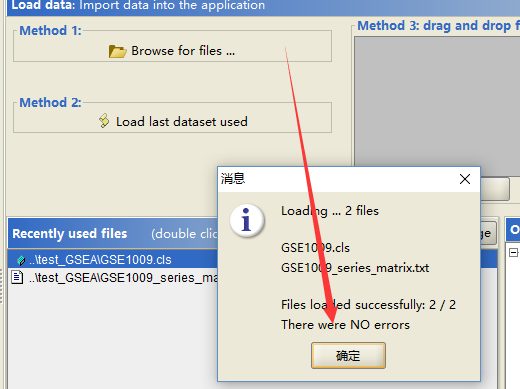
确定无误,就开始运行,运行需要设置一定的参数!

what's the output ?
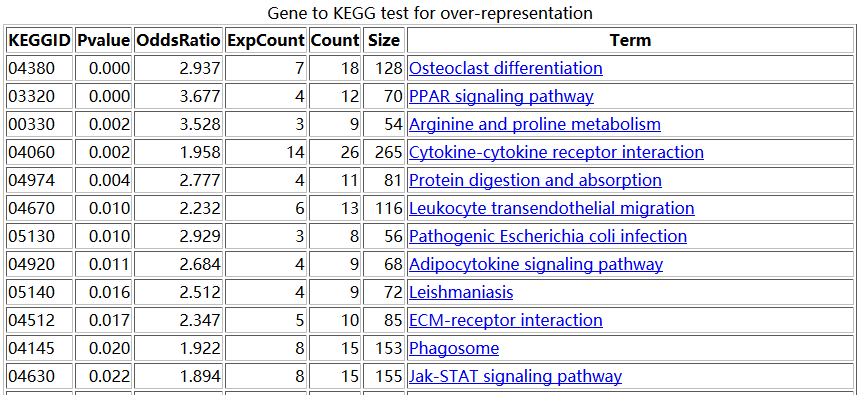
输出的数据非常多,对你选择的gene set数据集里面的每个set都会分析看看是否符合富集的标准,富集就出来一个报告。
点击success就能进入报告主页,里面的链接可以进入任意一个分报告。
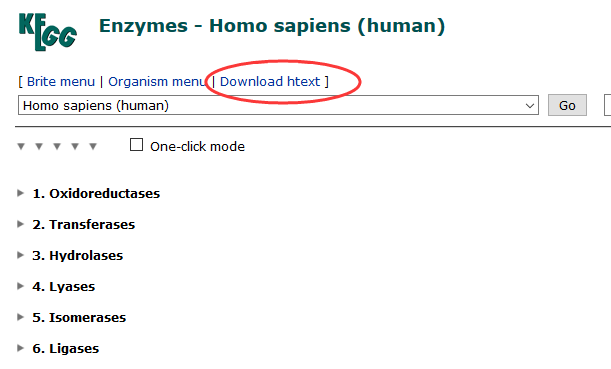
最大的特色是提供了大量的数据集:You can browse the MSigDB from the Molecular Signatures Database page of the GSEA web site or the Browse MSigDB page of the GSEA application. To browse the MSigDB from the GSEA application:
有些文献是基于GSEA的: