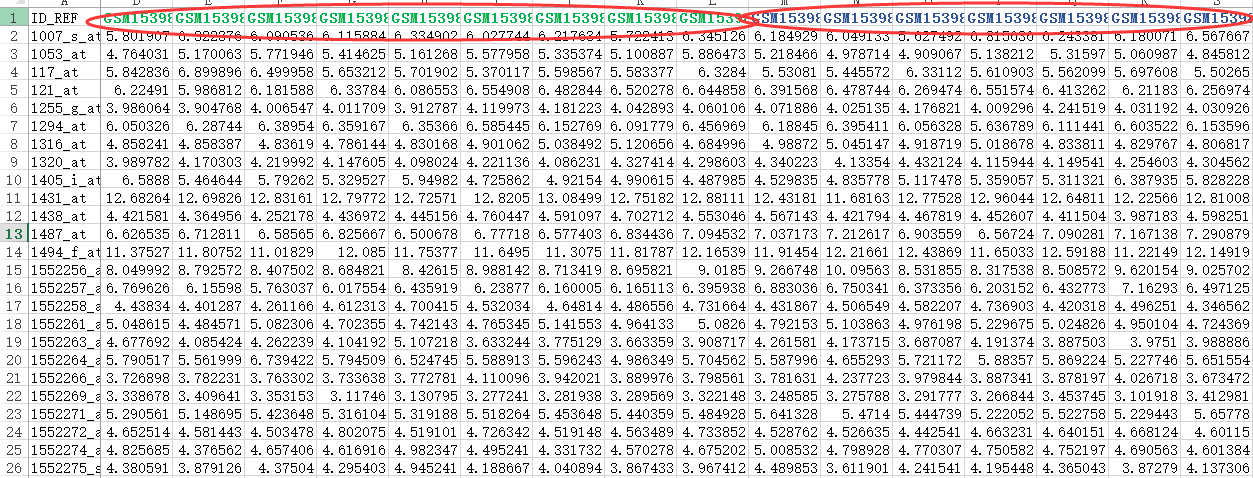
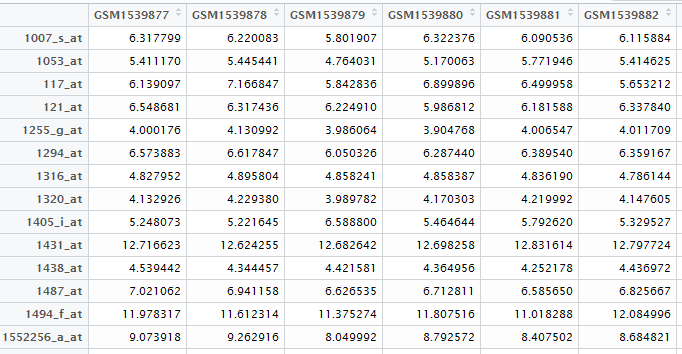
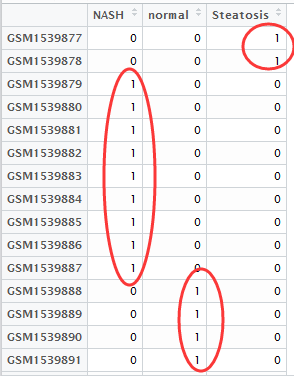
首先,我们要明白,limma接受的输入参数就是一个表达矩阵,而且是log后的表达矩阵(以2为底)。
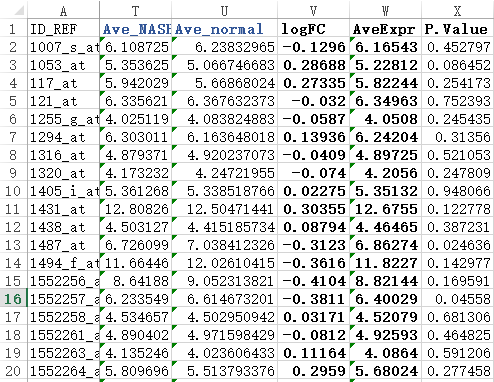
那么最后计算得到的logFC这一列的值,其实就是输入的表达矩阵中case一组的平均表达量减去control一组的平均表达量的值,那么就会有正负之分,代表了case相当于control组来说,该基因是上调还是下调。
我之前总是有疑问,明明是case一组的平均表达量和control一组的平均表达量差值呀,跟log foldchange没有什么关系呀。
后来,我终于想通了,因为我们输入的是log后的表达矩阵,那么case一组的平均表达量和control一组的平均表达量都是log了的,那么它们的差值其实就是log的foldchange
首先,我们要理解foldchange的意义,如果case是平均表达量是8,control是2,那么foldchange就是4,logFC就是2咯
那么在limma包里面,输入的时候case的平均表达量被log后是3,control是1,那么差值是2,就是说logFC就是2。
这不是巧合,只是一个很简单的数学公式log(x/y)=log(x)-log(y)