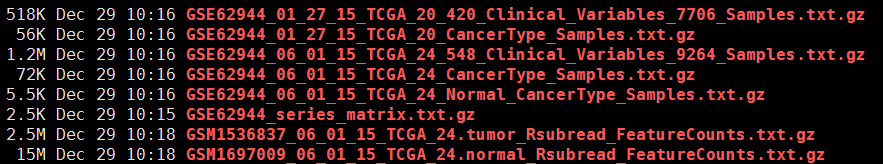
该软件是broad institute写的一个数据接口,主要是供他人下载TCGA的所有寄存在broad institute的免费数据,主要是level3,level4的数据。(说错了,好像只有level4的数据,就是可以发文章的分析结果及图片)
软件下载地址:https://confluence.broadinstitute.org/display/GDAC/Download
懂它的使用规则,编码规则即可:
就是一个很简单的shell脚本而已,根据几个用户自定义参数来选择性的下载数据。
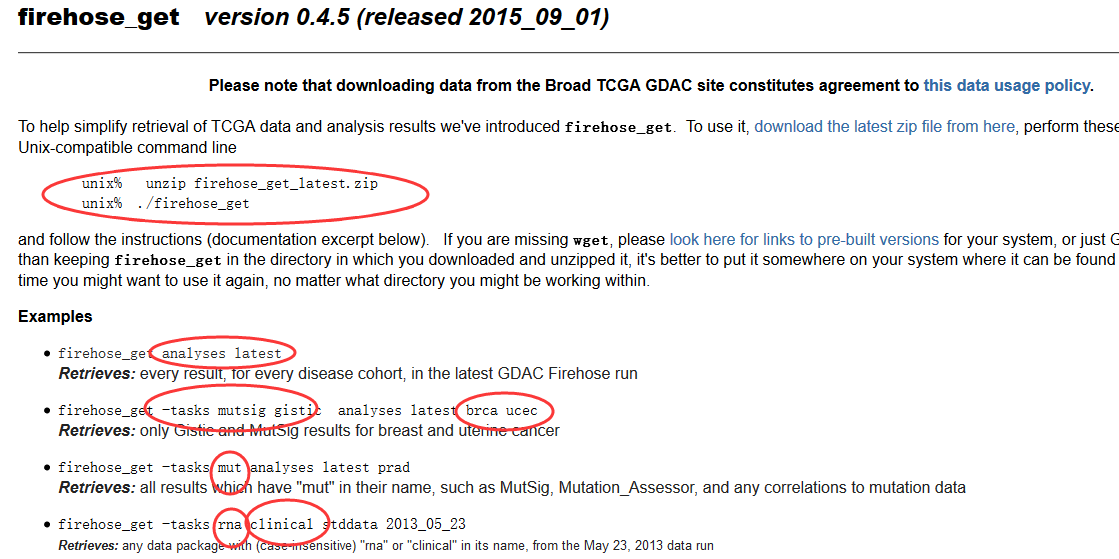
我们可以用-t这个参数来指定下载的数据类型,可以是mut/rna/mutsig/gistic等各种数据,至于这些单词代表什么意义,需要自己去看说明书啦
还可以指定时间,截止到什么时间的数据!
它支持的癌症种类:
ACC BLCA BRCA CESC COAD COADREAD DLBC ESCA GBM HNSC KICH KIRC KIRP LAML LGG LIHC LUAD LUSC OV PAAD PANCANCER PANCAN8 PANCAN12 PRAD READ SARC SKCM STAD THCA UCEC UCS
这些癌症种类的简称,也是可以去官网里面看到的!官网:http://gdac.broadinstitute.org