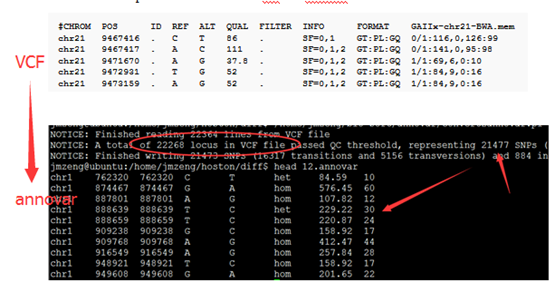
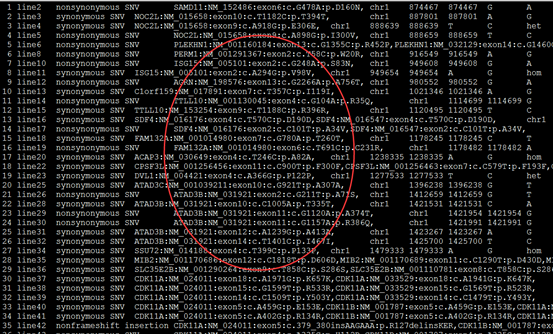
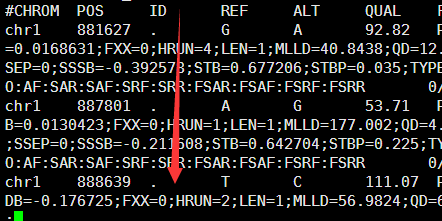
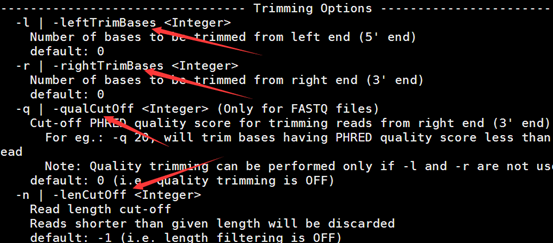
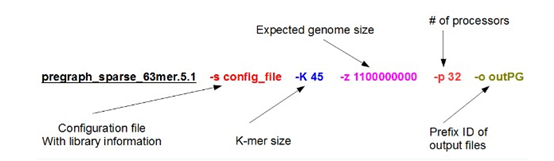
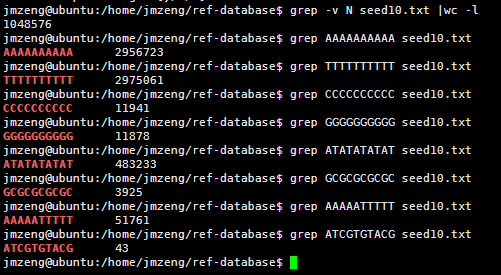
感觉大家对很多生物信息学的术语都不甚了解,我这里简单的从一个基因开始,扩展开来讲一讲生信数据库,及它相关的一些术语!
我要讲的基因是BRCA1,这是一个与乳腺癌以及卵巢癌都息息相关的基因。而BRCA1是它的英文缩写简称,也是通常学者们进行交流十它的名字。它的全称是breast cancer 1,每个基因都会有一个简称,比如下面这些,在human里面这些简称多大47732个,正常人都不会认识它们所有,只需要碰到了去数据库搜索即可,但是搞医疗健康的,必须熟悉癌症50基因。
这样的缩写简称其实弊端很多,单词毕竟是有限的,而且缩写也没有语义。所以NCBI给每个基因都定义了一个entrez ID号,是整数的排序,具体大家可以去看NCBI发的一篇文献,专门讲解了entrez ID号的好处。
1 A1BG
2 A2M
3 A2MP1
9 NAT1
10 NAT2
11 NATP
12 SERPINA3
13 AADAC
14 AAMP
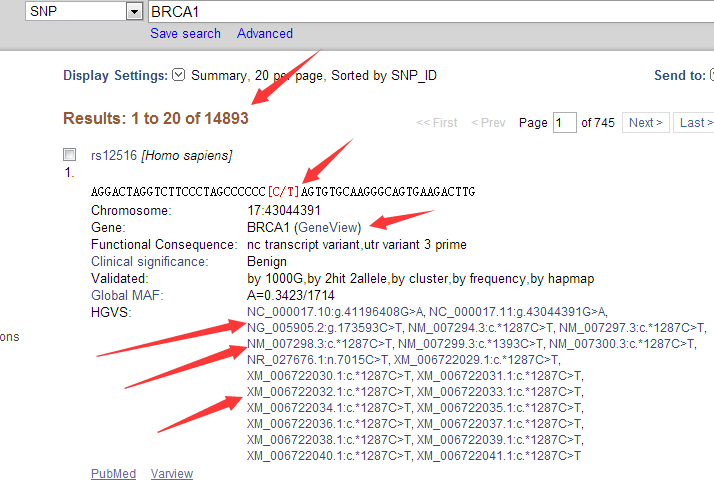
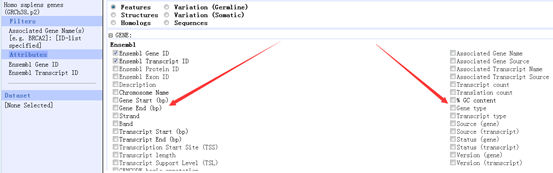
这里我们来找一下我们的BRCA1这个基因在生物信息数据库里面的其它信息,在NCBI的ftp里面有一个文件是Homo_sapiens.gene_info里面包含着人类所以基因的全部信息
9606 首先这个基因在human上面的,而human被NCBI定义的taxid是9606
672 然后这个基因的被NCBI定义的entrez ID号是672
BRCA1 这个当然就是这个基因的英文缩写名称啦
- 这个表明这个基因在负链什么
BRCAI|BRCC1|BROVCA1|FANCS|IRIS|PNCA4|PPP1R53|PSCP|RNF53
这个可能是基因以前的名称,或者是在其它研究领域的一些名称。MIM:113705|HGNC:HGNC:1100|Ensembl:ENSG00000012048|HPRD:00218|Vega:OTTHUMG00000157426
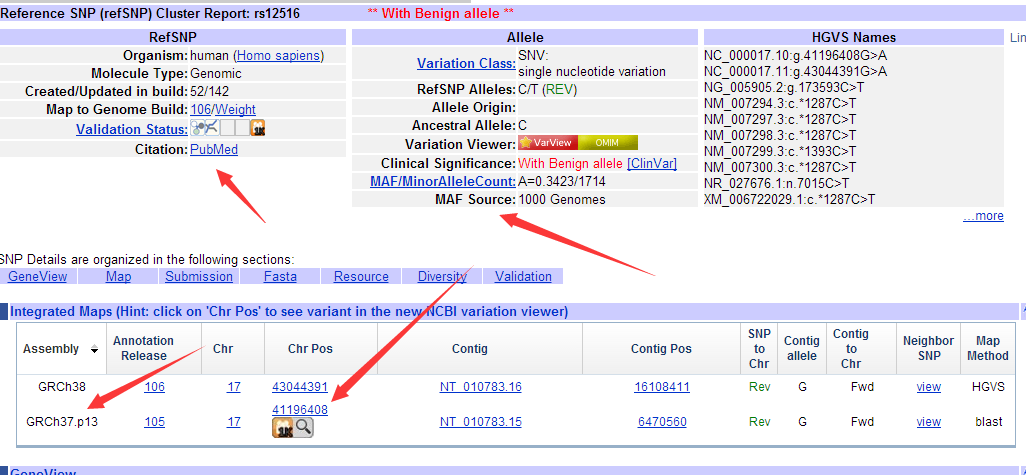
这里面包含在它在其它数据库的信息,我们的NCBI用entrez ID号672来标识它,相应的ensembl数据用ensembl ID号ENSG00000012048来标识它,还有什么MIM数据库,HGNC数据库,Vega数据库我就不详细讲啦
17 17q21 这个说明它在human的17号染色体的位置信息
下面一堆都是这个基因的描述,它的功能等等。
breast cancer 1, early onset protein-coding BRCA1 breast cancer 1, early onset
O BRCA1/BRCA2-containing complex, subunit 1|Fanconi anemia, complementation group S|RING finger protein 53|breast and ovarian cancer susceptibility protein 1|breast and ovarian cancer sususceptibility protein 1|breast cancer type 1 susceptibility protein|protein phosphatase 1, regulatory subunit 53
20150201
这样我们就把好几个数据库给串起来了,也大致了解了一个基因的各种信息,但是,这样肯定是不够的。
接下来我们就不用BRCA1来称呼这个基因了,我们统一用NCBI定义entrez ID号672来称呼这个基因,当然用ensembl ID号ENSG00000012048也可以,它们都是比较通用的。
ENSG00000012048 672 这个基因在GO数据库里面可以找到67个功能信息,分别是以下
GO:0000151 GO:0000724 GO:0000724 GO:0000794 GO:0003677 GO:0003684 GO:0003713 GO:0003723 GO:0004842 GO:0005515 GO:0005634 GO:0005654 GO:0005694 GO:0005737 GO:0005886 GO:0006260 GO:0006281 GO:0006301 GO:0006302 GO:0006302 GO:0006349 GO:0006357 GO:0006359 GO:0006633 GO:0006915 GO:0006974 GO:0006978 GO:0007059 GO:0007098 GO:0008270 GO:0008274 GO:0008630 GO:0009048 GO:0010212 GO:0010575 GO:0010628 GO:0015631 GO:0016567 GO:0016874 GO:0019899 GO:0030521 GO:0030529 GO:0031398 GO:0031436 GO:0031572 GO:0031625 GO:0035066 GO:0035067 GO:0042127 GO:0042981 GO:0043009 GO:0043234 GO:0043627 GO:0044030 GO:0044212 GO:0045717 GO:0045739 GO:0045766 GO:0045892 GO:0045893 GO:0045893 GO:0045944 GO:0045944 GO:0046600 GO:0050681 GO:0051571 GO:0051572 GO:0051573 GO:0051574 GO:0051865 GO:0070512 GO:0070531 GO:0071158 GO:0071356 GO:0071681 GO:0085020 GO:1902042 GO:2000378 GO:2000617 GO:2000620
由于GO太多了,我简单讲几个
ubiquitin ligase complex
double-strand break repair via homologous recombination
double-strand break repair via homologous recombination
condensed nuclear chromosome
DNA binding
damaged DNA binding
transcription coactivator activity
RNA binding
ubiquitin-protein transferase activity
protein binding
都是描述这个基因的功能的。
到这里我们大致了解了这个基因的功能,但是还不够。
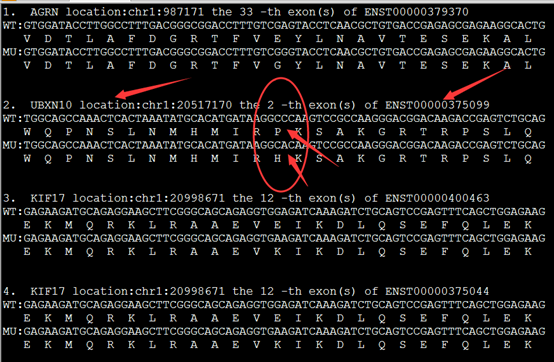
然后可以查到它有一下6个转录本,都有二十多个外显子。
NR_027676
NM_007300
NM_007299
NM_007298
NM_007297
NM_007294
在hg19这个参考基因组的起始终止坐标,还有各个外显子的起始终止坐标都能找到。
41196311,41199659,41201137,41203079,41209068,41215349,41215890,41219624,41222944,41226347,41228504,41234420,41242960,41243451,41247862,41249260,41251791,41256138,41256884,41258494,41267742,41276033,41277198
41197819,41199720,41201211,41203134,41209152,41215390,41215968,41219712,41223255,41226538,41228631,41234592,41243049,41246877,41247939,41249306,41251894,41256278,41256973,41258550,41267796,41276132,41277340
http://asia.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000012048;r=17:43044295-43125483
在ensembl里面关于这个基因的描述如下。
breast cancer 1, early onset [Source:HGNC Symbol;Acc:HGNC:1100]
BRCC1, FANCS, PPP1R53, RNF53
Chromosome 17: 43,044,295-43,125,483 reverse strand.
chromosome:GRCh38:CM000679.2:43044295:43125483:1
This gene has 29 transcripts (splice variants), 63 orthologues, is a member of 4 Ensembl protein families and is associated with 11 phenotypes.
RefSeq Gene ID 672
Uniprot identifiers: P38398
而且ensembl里面可以可视化这个基因的所有信息。
然后简单检索一下关于这个BRCA1基因的文献发表状况,居然多达2111篇文献,看来这个基因很火呀!!!
awk '{if ($1==9606 && $2==672) print }' gene2pubmed |wc
9606 672 1676470
9606 672 2001833
9606 672 2270482
9606 672 4506230
9606 672 7481765
9606 672 7545954
9606 672 7550349
9606 672 7795652
9606 672 7894491
9606 672 7894492
第三列1676470等编号是pubmed数据库的文献编号,可以直接找到关于这个基因的文献发表情况。
而直接在NCBI的pubmed数据库里面可以搜到多达11339篇文献。
esearch -db pubmed -query 'BRCA1'
Esearch这个程序是NCBI提供的,挺好用的,希望大家可以熟悉一下。
esearch -db pubmed -query 'BRCA1' | efetch -format docsum | xtract -pattern DocumentSummary -present Author -and Title -element Id -first "Author/Name" -element Title >BRCA1.pubmed
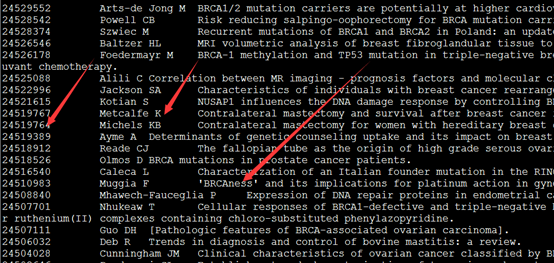
用这个代码,可以找到所有关于这个BRCA1基因的文献的作者及标题,这样可以统计在这个基因领域的研究者最出名的是谁。
至于这个基因的序列,及其转录本翻译的蛋白我就不列了,太长了,而且占位子