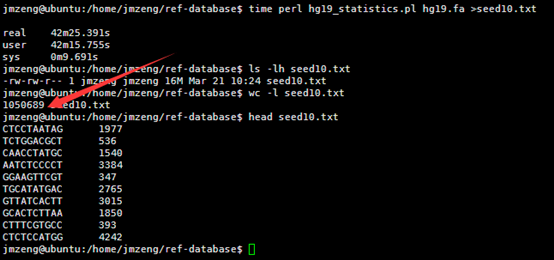
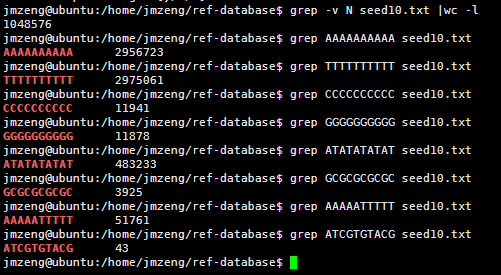
VCF是1000genome计划定义的测序比对突变说明文件,熟悉VCF文件的都知道,第三列是ID号,也就是说对该突变在dbsnp的数据库的编号。大多时候都是用点号占位,代表不知道在dbsnp的数据库的编号,这时候就需要我们自己来注释了。
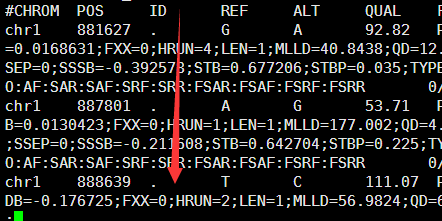
其实,这是一个非常简单的事情,因为有了CHROM和pos,只要找到一个文件,就可以自己写脚本来映射到它的ID号,但是找这个文件比较困难,我也是搜索了好久才找到的。
http://varianttools.sourceforge.net/Annotation/DbSNP
这里面提到了最新版的数据库是dbSNP138
The default version of our dbSNP annotation is currently referring to dbSNP138 (using hg19 coordinates) as shown below. However, users can also retrieve older versions of dbSNP: db135, dbSNP129, dbSNP130, dbSNP131 and dbSNP132. The 129 and 130 versions use hg18 as a reference genome and 131, 132, 135 and later use hg19. The archived versions can be used by a variant tools project by referring to their specific names - for example: dbSNP-hg18_129.
所以我就换了关键词,终于搜的了
http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?view+summary=view+summary&build_id=138
http://asia.ensembl.org/info/genome/variation/sources_documentation.html?redirect=no
SNP 138 database (232,952,851 million altogether).

有一个bioconductor包是专门来做snp过滤的
http://www.bioconductor.org/packages/release/bioc/html/VariantAnnotation.html
首先下载vcf文件。
nohup wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/00-All.vcf.gz &
这个文件很大,解压开是
如果大家对snp不了解,可以去查看它的各种介绍以及分类
http://moma.ki.au.dk/genome-mirror/cgi-bin/hgTrackUi?db=hg19&g=snp138
其实我这里本来有个hg19_snp141.txt文件,如下
1 10020 A - .
1 10108 C T .
1 10109 A T .
1 10139 A T .
1 10145 A - .
1 10147 C - .
1 10150 C T .
1 10177 A C .
1 10180 T C .
1 10229 A - .
还可以下载一些文件,如bed_chr_1.bed
chr1 175292542 175292543 rs171 0 -
chr1 20542967 20542968 rs242 0 +
chr1 6100897 6100898 rs538 0 -
chr1 93151988 93151989 rs546 0 +
chr1 15220328 15220329 rs549 0 +
chr1 203744004 203744005 rs568 0 +
chr1 23854550 23854551 rs665 0 -
chr1 53213656 53213657 rs672 0 +
chr1 173907422 173907423 rs677 0 -
当然还有那个ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/00-All.vcf.gz 18G的文件,是VCF格式
##fileformat=VCFv4.0
##fileDate=20150218
##source=dbSNP
##dbSNP_BUILD_ID=142
##reference=GRCh38
#CHROM POS ID REF ALT QUAL FILTER INFO
1 10108 rs62651026 C T . . RS=62651026;RSPOS=10108;dbSNPBuildID=129;SSR=0;SAO=0;VP=0x050000020005000002000100;WGT=1;VC=SNV;R5;ASP
所以这个文件就是我们想要的最佳文件,提取前三列就够啦
#CHROM POS ID
1 10108 rs62651026
1 10109 rs376007522
1 10139 rs368469931
1 10144 rs144773400
1 10150 rs371194064
1 10177 rs201752861
1 10177 rs367896724
1 10180 rs201694901
1 10228 rs143255646
1 10228 rs200462216
这样就可以通过脚本用hash把我们自己找到的hash跟数据库的rs编号对应起来啦