估计很多小伙伴都没有听过sepsis,现在翻译成中文是脓毒病,很多人会把它与那个缺乏维他命C的败血症混淆,其实完全不一样,因为sepsis致死率非常高!
| sepsis | 英[ˈsepsɪs] | 美['sepsɪs] |
| n. | 脓毒病; 脓毒疾; |
[例句]This may be of value in the treatment of meningitis and sepsis.
这可能会在治疗脑膜炎和败血症上有一定价值。
估计很多小伙伴都没有听过sepsis,现在翻译成中文是脓毒病,很多人会把它与那个缺乏维他命C的败血症混淆,其实完全不一样,因为sepsis致死率非常高!
| sepsis | 英[ˈsepsɪs] | 美['sepsɪs] |
| n. | 脓毒病; 脓毒疾; |
无意中看到了这个网站,比wiki的还有全面和专业。搜集了现有还算比较出名的比对软件,并且列出来了,还做了简单评价,里面对比对工具的收集,主要是基于2012年的一个综述《Tools for mapping high-throughput sequencing data》,相信应该是有不少人都看过这篇综述的,其实生物信息初学者应该自己去文献数据库找点感兴趣的关键词的综述多看看,广泛涉猎总没有坏处的。
<img src="http://www.ebi.ac.uk/~nf/hts_mappers/mappers_timeline.jpeg" alt="Mappers Timeline" width="800">
The following Table enables a comparison of mappers based on different characteristics. The table can be sorted by column (just click on the column name). The data was collected from different sources and in some cases was provided by the developers. For execution times and memory requirements we refer to the above mentioned review (supplementary data is available here).
这个软件其实我真心不需要讲些什么了,它的官网写的太好了,简直就是软件说明书的典范
http://www.nipgr.res.in/ngsqctoolkit.html
它列出了它的几个功能模块,还给出了下载地址,还给出了说明文档,下载压缩包,解压即可使用啦
更重要的是给出了测试数据和测试的结果,而且还专门测试了不同测序平台及不同的测序策略的使用说明
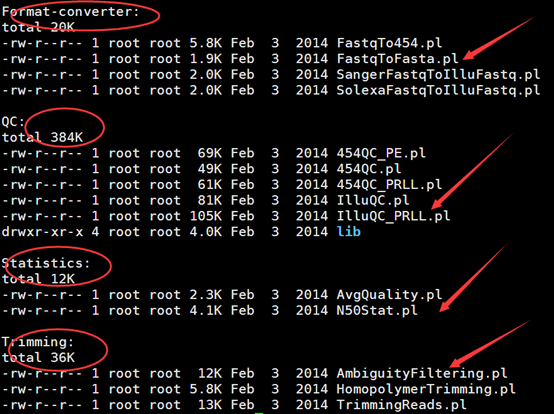
里面就是一些perl测序,其实自己都可以写的,分成了四大类。
其中统计的那个平均测序质量,我在前面仿写fastqc就写过,至于那个统计N50,更是生信常用的脚本。
但是大家可以看看这个perl程序来学perl语言,蛮不错的这些程序,都写的很标准。
比如那个TrimmingReads.pl
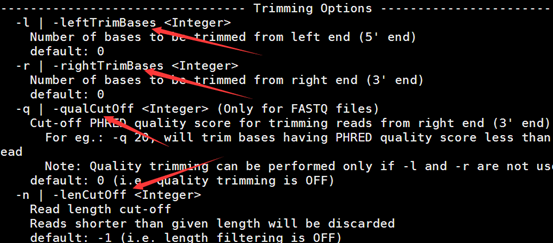
可以根据四个参数来选择性的对我们的原始reads进行过滤,当然很多其它的程序也有类似的功能,它的参数分别是铲掉5端的几个碱基或者3端的,或者根据测序质量来切除碱基,或者根据reads长度来取舍,都是挺实用的功能。但是我一般用LengthSort和DynamicTrim那两个程序,原因很简单,我老师是这样用的,所以我习惯了,哈哈