由于之前就简单的看了看bowtie作者的ppt,没有完全吃透就开始敲代码了,写了十几个程序最后我自己都搞不清楚进展到哪一步了,所以我现在整理一下,从新开始!!!
首先,bowtie的作用就是在一个大字符串里面搜索一个小字符串!那么本身就有一个非常笨的复杂方法来搜索,比如,大字符串长度为100万,小字符串为10,那么就依次取出大字符串的10个字符来跟小字符串比较即可,这样的算法是非常不经济的,我简单用perl代码实现一下。
[perl]
#首先读取大字符串的fasta文件
open FH ,"<$ARGV[0]";
$i=0;
while (<FH>) {
next if /^>/;
chomp;
$a.=(uc);
}
#print "$a\n";
#然后接受我们的小的查询字符串
$query=uc $ARGV[1];
$len=length $a;
$len_query=length $query;
$a=$a.'$'.$a;
#然后依次循环取大字符串来精确比较!
foreach (0..$len-1){
if (substr($a,$_,$len_query) eq $query){
print "$_\n";
#last;
}
}
[/perl]
这样在时间复杂度非常恐怖,尤其是对人的30亿碱基。
正是因为这样的查询效率非常低,所以我们才需要用bwt算法来构建索引,然后根据tally来进行查询
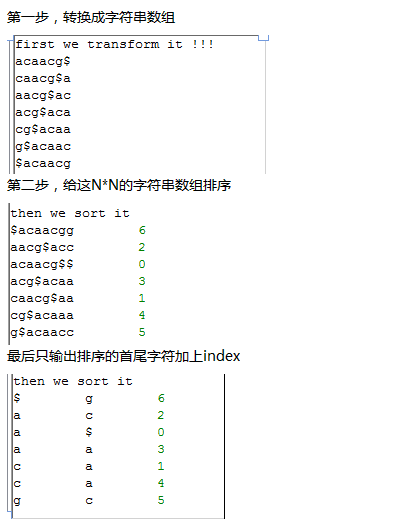
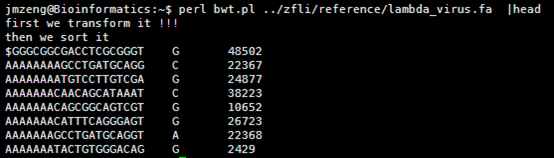
其中构建索引有三种方式,我首先讲最效率最低的那种索引构造算法,就是依次取字符串进行旋转,然后排序即可。
[perl]
$a=uc $ARGV[0];
$len=length $a;
$a=$a.'$'.$a;
foreach (0..$len){
$hash{substr($a,$_,$len+1)}=$_;
}
#print "$_\t$hash{$_}\n" foreach sort keys %hash;
print substr($_,-1),"\t$hash{$_}\n" foreach sort keys %hash;
[/perl]
这个算法从时间复杂度来讲是非常经济的,对小字符串都是瞬间搞定!!!
perl rotation_one_by_one.pl atgcgtanngtc 这个字符串的BWT矩阵索引如下!
C 12
T 6
$ 0
T 11
G 3
T 2
C 4
N 9
N 8
A 7
G 5
G 10
A 1
但同样的,它也有一个无法避免的弊端,就是内存消耗太恐怖。对于30亿的人类碱基来说,这样旋转会生成30亿乘以30亿的大矩阵,一般的服务器根本hold不住的。
最后我讲一下,这个BWT矩阵索引如何还原成原字符串,这个没有算法的差别,因为就是很简单的原理。
[perl]
#first read the tally !!!
#首先读取上面输出的BWT矩阵索引文件。
open FH,"<$ARGV[0]";
$hash_count{'A'}=0;
$hash_count{'C'}=0;
$hash_count{'G'}=0;
$hash_count{'T'}=0;
while(<FH>){
chomp;
@F=split;
$hash_count{$F[0]}++;
$hash{$.}="$F[0]\t$F[1]\t$hash_count{$F[0]}";
#print "$hash{$.}\n";
}
$all_a=$hash_count{'A'};
$all_c=$hash_count{'C'};
$all_g=$hash_count{'G'};
$all_t=$hash_count{'T'};
$all_n=$hash_count{'N'};
#start from the first char !
$raw='';
&restore(1);
sub restore{
my($num)=@_;
my @F=split/\t/,$hash{$num};
$raw.=$F[0];
my $before=$F[0];
if ($before eq 'A') {
$new=$F[2]+1;
}
elsif ($before eq 'C') {
$new=1+$all_a+$F[2];
}
elsif ($before eq 'G') {
$new=1+$all_a+$all_c+$F[2];
}
elsif ($before eq 'N') {
$new =1+$all_a+$all_c+$all_g+$F[2];
}
elsif ($before eq 'T') {
$new=1+$all_a+$all_c+$all_g+$all_n+$F[2];
}
elsif ($before eq '$') {
chop $raw;
$raw = reverse $raw;
print "$raw\n";
exit;
}
else {die "error !!! we just need A T C N G !!!\n"}
#print "$F[0]\t$new\n";
&restore($new);
}
[/perl]