我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
library(DESeq2)
library(limma)
library(pasilla)
data(pasillaGenes)
exprSet=counts(pasillaGenes) ##做好表达矩阵
group_list=pasillaGenes$condition##做好分组因子即可(colData <- data.frame(row.names=colnames(exprSet), group_list=group_list))
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)##上面是第一步第一步,构建dds这个对象,需要一个表达矩阵和分组矩阵!!!
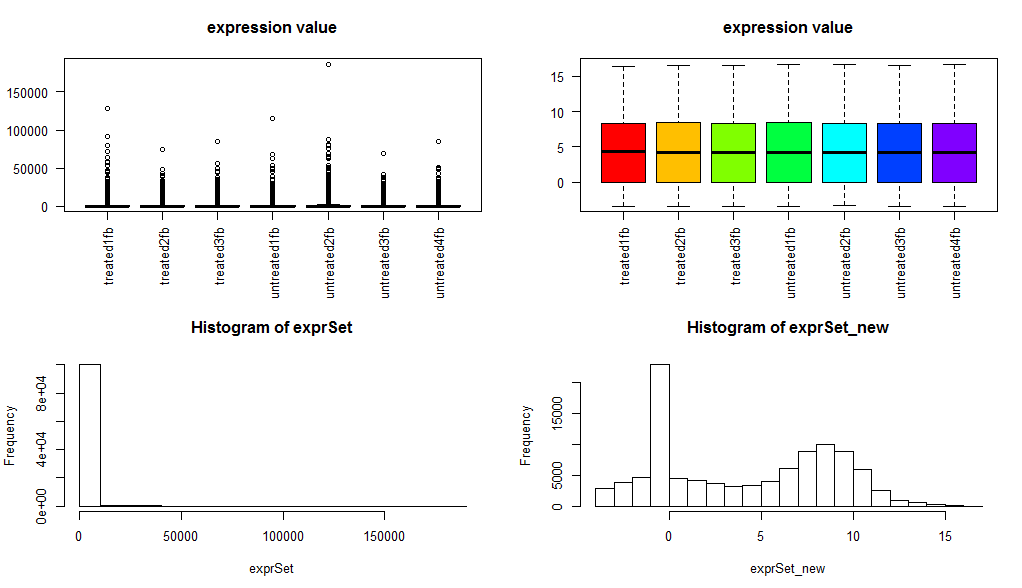
dds2 <- DESeq(dds) ##第二步,直接用DESeq函数即可resultsNames(dds2)res <- results(dds2, contrast=c("group_list","treated","untreated"))## 提取你想要的差异分析结果,我们这里是treated组对untreated组进行比较resOrdered <- res[order(res$padj),]resOrdered=as.data.frame(resOrdered)可以看到程序非常好用!它只对RNA-seq的基因的reads的counts数进行分析,请不要用RPKM等经过了normlization的表达矩阵来分析。值得一提的是DESeq2软件独有的normlization方法!rld <- rlogTransformation(dds2) ## 得到经过DESeq2软件normlization的表达矩阵!
exprSet_new=assay(rld)
par(cex = 0.7)
n.sample=ncol(exprSet)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
par(mfrow=c(2,2))
boxplot(exprSet, col = cols,main="expression value",las=2)
boxplot(exprSet_new, col = cols,main="expression value",las=2)
hist(exprSet)
hist(exprSet_new)