BayesPeak也是peaks caller家族一员,用的人也不少,我这次也试了一下,因为是R的bioconductor系列包,所以直接在R里面安装就好,但是有几个点需要注意,我比对的基因组不只是Chr1~22,X,Y,M,还有一些contig和scaffold,需要在bam文件里面去除的,而且BayesPeak比较支持读取BED文件,可以直接转为GRanges对象,虽然它号称可以使用多核,但是计算速度还是非常慢。 Continue reading
七
05
BayesPeak也是peaks caller家族一员,用的人也不少,我这次也试了一下,因为是R的bioconductor系列包,所以直接在R里面安装就好,但是有几个点需要注意,我比对的基因组不只是Chr1~22,X,Y,M,还有一些contig和scaffold,需要在bam文件里面去除的,而且BayesPeak比较支持读取BED文件,可以直接转为GRanges对象,虽然它号称可以使用多核,但是计算速度还是非常慢。 Continue reading
此文专门讲这个软件如何用,但是跟我以前写的软件说明书又不大一样,主要是因为我用MACS2这个软件call peaks并没有达到预期的结果,所以就多使用了几个软件,其中PeakRanger尤其值得一提,安装特别简单,而且处理数据的速度特别快,结果也非常容易理解,更重要的是它给出一个网页版的报告,里面有所有找到的符合要求的peaks的可视化图片!!!! Continue reading
CHIP-seq测序的本质还是目标片段捕获测序,跟WES不同的是,它不是通过固定的芯片探针来固定的捕获基因组上面特定序列,而是根据你选择的IP不同,你细胞或者机体状态不同,捕获到的序列差异很大!而我们研究的重点,就是捕获到的差异。而我们对CHIP-seq测序数据寻找peaks的本质就是得到所有测序数据比对在全基因组之后在正个基因组上面的测序深度里面寻找比较突出的。比如对WES数据来说,各个外显子,或者外显子的5端到3端,理论上测序深度应该是一致的,都是50X~200X,画一个测序深度曲线,应该是近似于一条直线。对我们的CHIP-seq测序数据来说,在所捕获的区域上面,理论上测序深度是绝对不一样的,应该是近似于一个山峰。而那些覆盖度高的地方,山顶,就是我们的IP所结合的热点,也就是我们想要找的peaks,在IGV里面看到大致是下面这样:
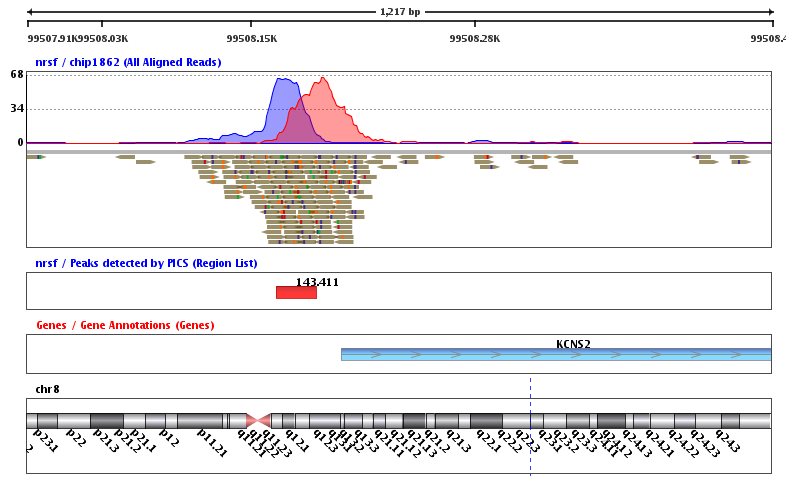
可以看到测序的reads分布是绝对的不均匀的!我们通常说的CHIP-seq测序的IP,可以是各个组蛋白的各个修饰位点对应的抗体,或者是各种转录因子的抗体,等等
如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的read所覆盖(我们测的是一个细胞群体,read出现次数多,说明该位置被TF结合的几率大)。那么,read数达到多少才叫多?这就要用到统计检验喽。假设TF在基因组上的分布是没有任何规律的,那么,测序得到的read在基因组上的分布也必然是随机的,某个碱基上覆盖的read的数目应该服从二项分布。
具体统计学原理直接看原创吧:http://www.plob.org/2014/05/08/7227.html
比对本质是是很简单的了,各种mapping工具层出不穷,我们一般常用的就是BWA和bowtie了,我这里就挑选bowtie2吧,反正别人已经做好了各种工具效果差异的比较,我们直接用就好了,代码如下:
## step5 : alignment to hg19/ using bowtie2 to do alignment
## ~/biosoft/bowtie/bowtie2-2.2.9/bowtie2-build ~/biosoft/bowtie/hg19_index /hg19.fa ~/biosoft/bowtie/hg19_index/hg19
## cat >run_bowtie2.sh
ls *.fastq | while read id ;
do
echo $id
#~/biosoft/bowtie/bowtie2-2.2.9/bowtie2 -p 8 -x ~/biosoft/bowtie/hg19_index/hg19 -U $id -S ${id%%.*}.sam 2>${id%%.*}.align.log;
#samtools view -bhS -q 30 ${id%%.*}.sam > ${id%%.*}.bam ## -F 1548 https://broadinstitute.github.io/picard/explain-flags.html
# -F 0x4 remove the reads that didn't match
samtools sort ${id%%.*}.bam ${id%%.*}.sort ## prefix for the output
# samtools view -bhS a.sam | samtools sort -o - ./ > a.bam
samtools index ${id%%.*}.sorted.bam
done
这个索引~/biosoft/bowtie/hg19_index/hg19需要自己提取建立好,见前文
初步比对的sam文件到底该如何过滤,我查了很多文章都没有给出个子丑寅卯,各执一词,我也没办法给大家一个标准,反正我测试了好几种,看起来call peaks的差异不大,就是得不到文章给出的那些结果!!
一般来说,初步比对的sam文件只能选取unique mapping的结果,所以我用了#samtools view -bhS -q 30,但是结果并没什么改变,有人说是peak caller这些工具本身就会做这件事,所以取决于你下游分析所选择的工具。
给大家看比对的日志吧:
SRR1042593.fastq
16902907 reads; of these:
16902907 (100.00%) were unpaired; of these:
667998 (3.95%) aligned 0 times
12467095 (73.76%) aligned exactly 1 time
3767814 (22.29%) aligned >1 times
96.05% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042594.fastq
60609833 reads; of these:
60609833 (100.00%) were unpaired; of these:
9165487 (15.12%) aligned 0 times
39360173 (64.94%) aligned exactly 1 time
12084173 (19.94%) aligned >1 times
84.88% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042595.fastq
14603295 reads; of these:
14603295 (100.00%) were unpaired; of these:
918028 (6.29%) aligned 0 times
10403045 (71.24%) aligned exactly 1 time
3282222 (22.48%) aligned >1 times
93.71% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042596.fastq
65911151 reads; of these:
65911151 (100.00%) were unpaired; of these:
10561790 (16.02%) aligned 0 times
42271498 (64.13%) aligned exactly 1 time
13077863 (19.84%) aligned >1 times
83.98% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042597.fastq
22210858 reads; of these:
22210858 (100.00%) were unpaired; of these:
1779568 (8.01%) aligned 0 times
15815218 (71.20%) aligned exactly 1 time
4616072 (20.78%) aligned >1 times
91.99% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042598.fastq
58068816 reads; of these:
58068816 (100.00%) were unpaired; of these:
8433671 (14.52%) aligned 0 times
37527468 (64.63%) aligned exactly 1 time
12107677 (20.85%) aligned >1 times
85.48% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042599.fastq
24019489 reads; of these:
24019489 (100.00%) were unpaired; of these:
1411095 (5.87%) aligned 0 times
17528479 (72.98%) aligned exactly 1 time
5079915 (21.15%) aligned >1 times
94.13% overall alignment rate
[samopen] SAM header is present: 93 sequences.
SRR1042600.fastq
76361026 reads; of these:
76361026 (100.00%) were unpaired; of these:
8442054 (11.06%) aligned 0 times
50918615 (66.68%) aligned exactly 1 time
17000357 (22.26%) aligned >1 times
88.94% overall alignment rate
[samopen] SAM header is present: 93 sequences.
可以看到比对非常成功!!!我这里就不用表格的形式来展现了,毕竟我又不是给客户写报告,大家就将就着看吧。
博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
## step1 : download raw datacd ~mkdir CHIPseq_test && cd CHIPseq_testmkdir rawData && cd rawData## batch download the raw data by shell script :for ((i=593;i<601;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP033/SRP033492/SRR1042$i/SRR1042$i.sra;done
621M Jun 27 14:03 SRR1042593.sra (16.9M reads)2.2G Jun 27 15:58 SRR1042594.sra (60.6M reads)541M Jun 27 16:26 SRR1042595.sra (14.6M reads)2.4G Jun 27 18:24 SRR1042596.sra (65.9M reads)814M Jun 27 18:59 SRR1042597.sra (22.2M reads)2.1G Jun 27 20:30 SRR1042598.sra (58.1M reads)883M Jun 27 21:08 SRR1042599.sra (24.0M reads)2.8G Jun 28 11:53 SRR1042600.sra (76.4M reads)
## step2 : change sra data to fastq files.## cell line: MCF7 // Illumina HiSeq 2000 // 50bp // Single ends // phred+33ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump $id;donerm *sra
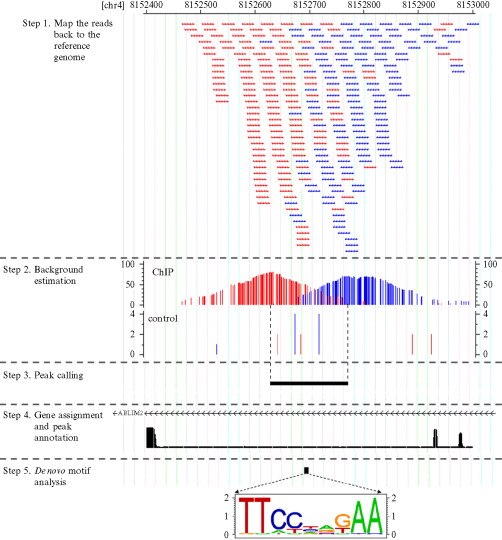
因为最近在研究CHIP-seq测序数据处理,发现有些文章重点并不是数据处理本身,而是对生物学基础知识的掌控以及实验设计,这篇文章我重点推荐一下,我略微翻译了一些,笔记如下: