我们分享过单细胞转录组下游的降维聚类分群的很多例子,比如:[人人都能学会的单细胞聚类分群注释] Continue reading
六
25
我们分享过单细胞转录组下游的降维聚类分群的很多例子,比如:[人人都能学会的单细胞聚类分群注释] Continue reading
最近有粉丝在我b站的数据挖掘视频课程发弹幕吐槽我授课时候作为例子的火山图不怎么好看,希望我提高一下自己的神秘,课程是:三年前的数据挖掘课程(TNBC表达矩阵探索) Continue reading
最近在有粉丝求助,他的scATAC-seq数据的分析,使用 scATAC pro 这个软件得到 cluster 里面的细胞类型好少。 Continue reading
看到一个临床wgs应用的文献速递,《Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers》,链接是:https://pubmed.ncbi.nlm.nih.gov/33704937/ Continue reading
最近在更新一个R包的时候,发生了如下所示的报错:
一个学徒跟着我做了七十多个转录组项目了,但是一直不能理解,凭什么这样的高通量筛选就能定位到具体的一两个基因。 Continue reading
是我太年轻
学员群有咨询 Agilent-038314 CBC Homo sapiens lncRNA + mRNA microarray V2.0 这个表达量芯片的数据处理问题,当然了,主要是芯片的探针ID对应基因名字的问题。 链接是; Continue reading
前面我带领大家通过IMGT数据库认知免疫组库,而且也一起[从IMGT数据库下载免疫组库相关fasta序列] Continue reading
最近在整理新冠疫情相关的单细胞文章,尤其是那些提供了数据集的而且还有配套GitHub代码的,超级棒的学习资料。 Continue reading
今天在《共享服务器第27个群》看到有粉丝提问,说他跑cibersort的时候,R代码运行超级慢,需要一些加速技巧。 Continue reading
看到我最近在报道一些生物信息学数据分析的吐槽点,见:
最近看到朋友圈转发的一大批“神医”做出违背祖宗的决定!
腾讯视频链接:https://v.qq.com/x/page/x3230xgj0x6.html
让我动容,敬佩之外我也想效仿一二,把我“祖传的”生物信息学技能公之于众,敞开门让大家学!我已经把这些技能录制成为了视频,并且在B站免费发布,已经组建了微信交流群的有下面这些:
当然了,如果你直接上手这些NGS组学数据分析实战有困难,说明你的Linux基础或者R语言不过关,也可以看c超级基础的内容:

再怎么强调生物信息学数据分析学习过程的计算机基础知识的打磨都不为过,我把它粗略的分成基于R语言的统计可视化,以及基于Linux的NGS数据处理:
把R的知识点路线图搞定,如下:
Linux的6个阶段也跨越过去 ,一般来说,每个阶段都需要至少一天以上的学习:
今天要介绍的文章是:Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019 ,它的数据链接是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110746 Continue reading
前面我们在推文 细胞亚群的特异性标记基因也许真的很难提到的Cancer-associated fibroblasts (CAFs)是比较难以精确的细分亚群。而且我们讨论了[T细胞可以简单分成4类] Continue reading
最近实验室同学在组会上分享了一篇很有意思的文章,是于 January 2021, 发表在CELL杂志的文章《Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer》,链接是:https://doi.org/10.1016/j.cell.2020.12.031 Continue reading
于2021年3月发表在CELL杂志的文章, 标题是:《In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer》,链接是:https://doi.org/10.1016/j.cell.2021.02.019 Continue reading
我在《生信菜鸟团》的一个推文 单细胞门户网站哪个更齐全,提到了生物信息学资源基本上都是欧洲的EBI的sanger研究所和美国的MIT的broad研究所创造和整理,单细胞领域也不例外。 Continue reading
前面我在《生信菜鸟团》公众号介绍了 小鼠的肿瘤免疫微环境推断可以用seq-ImmuCC,本来是想布置作业让大家下载https://www.ncbi.nlm.nih.gov/bioproject/PRJNA489661/ 的数据,自己走一波seq-ImmuCC实战。
但是发现这个并不是《生信技能树》公众号,所以我没办法布置练习题额,所以现在在《生信技能树》公众号再来一次哈!前面的例子:人人都能学会的单细胞聚类分群注释 ,第一次分群就非常漂亮!可以看到这个数据集GSE129516里面的6个样品都是有不同的免疫细胞亚群的,而且既然已经是有了降维聚类分群结果,就可以算出各个细胞亚群的比例啦!
我们的作业是,把GSE129516里面的6个样品的单细胞表达量矩阵简单的累加成为一个假的bulk表达量矩阵, 然后拿这个表达量矩阵去进行seq-ImmuCC实战,推断小鼠的肿瘤免疫微环境,就是各个免疫细胞比例。然后跟单细胞的真实免疫细胞各个亚群比例进行对比,测试一下seq-ImmuCC这个网页工具的表现情况!
苏州系统医学研究所苏吴爱平教授和秦晓峰教授合作,发布了一个基于转录组测序(RNA-Seq)数据对小鼠组织中10种主要免疫细胞组分进行预测的计算模型(seq-ImmuCC),将为测序数据提供免疫细胞层面的解读视角。相关研究以“seq-ImmuCC: Cell-CentricView of Tissue Transcriptome Measuring Cellular Compositions of ImmuneMicroenvironment From Mouse RNA-Seq Data”为题,于2018年6月发表于国际期刊Frontiersin Immunology。
吴爱平教授的课题组已经成功开发了两个计算模型,从组织样本的芯片表达谱。
在:http://218.4.234.74:3200/immune/manual
我看了看它的示例文件,很简单一个csv,如下所示:
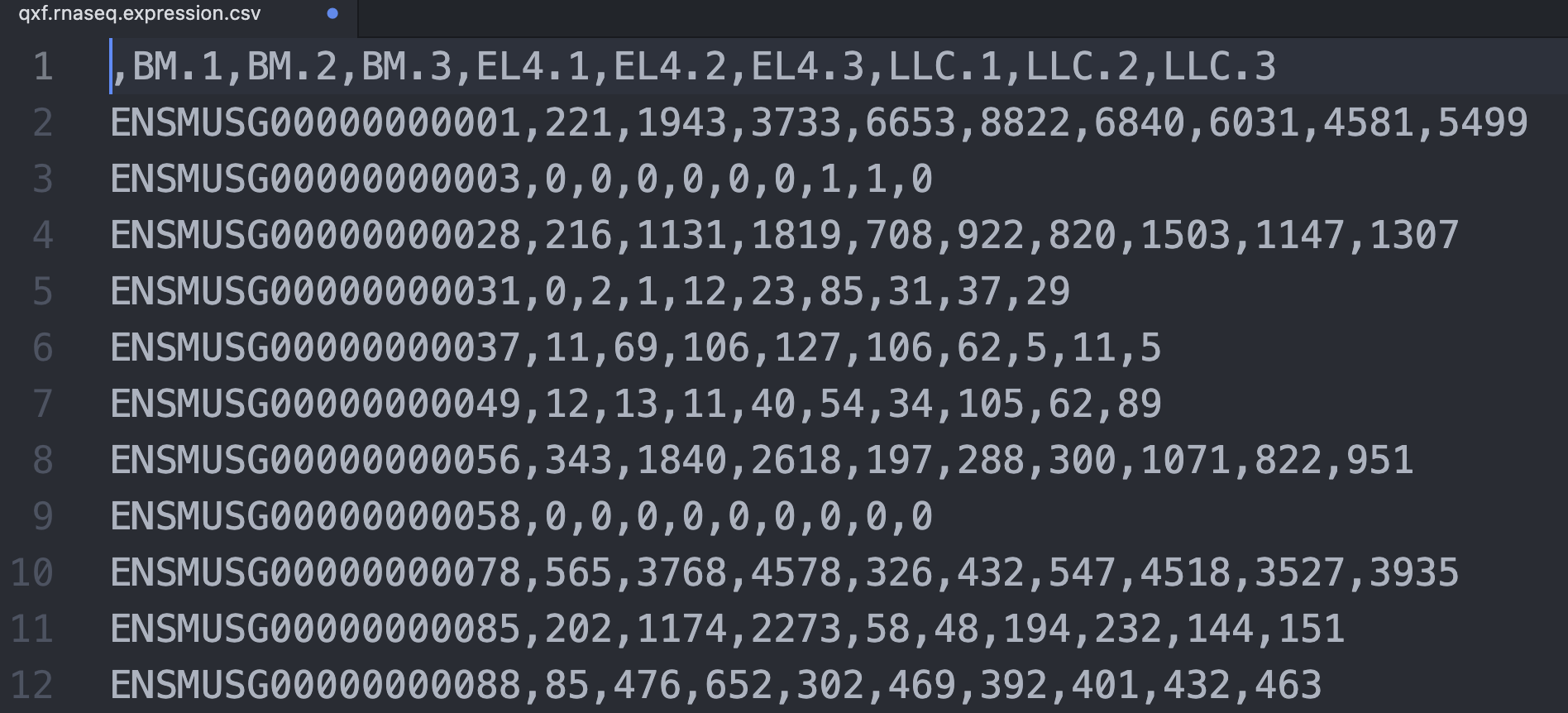
见:http://218.4.234.74:3200/immune/
也是很容易理解的结果,一个矩阵一个条形图,如下所示:
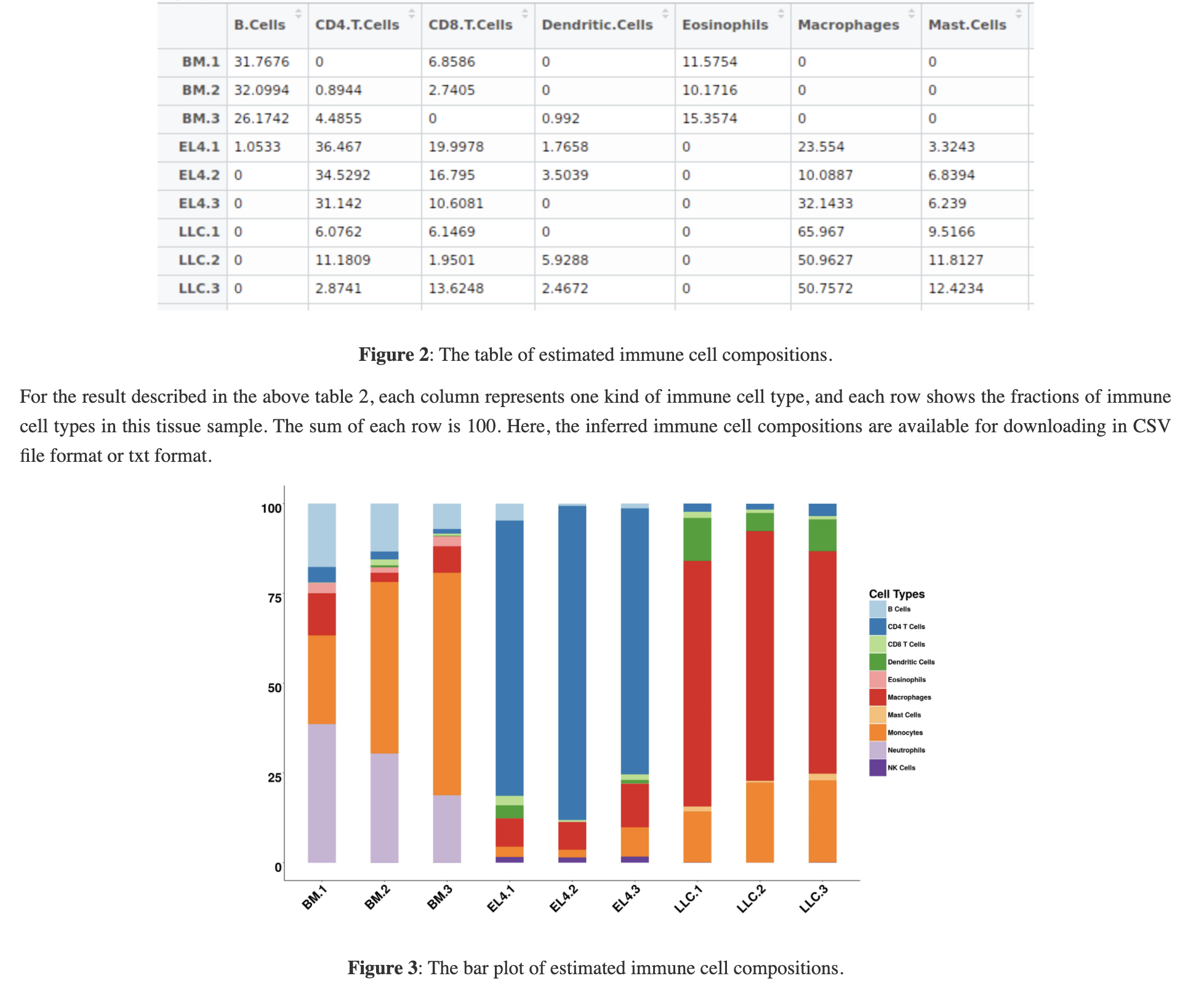
确实值得表扬,我测试了三次,体验都很好!仅仅是一个表达量矩阵的csv文件即可。