有很多读者来信,CHIP-seq数据比对后的bam文件如果根据基因组的所有基因来画热图,profile图呢?
十二
15
这本是我为论坛的基础板块写的一个基础知识点,但是浏览量实在有限,不忍它蒙尘,特在博客重新发布一次!原帖见:http://www.biotrainee.com/thread-511-1-1.html
gene symbol 是非常官方的,由HUGO 组织负责维护,有专门的数据库HGNC database of human gene names | HUGO
以前分析数据的时候,有一些基因的symbol很奇怪,让我百思不得其解,比如
C orf 系列基因,
HS.系列基因,
KRTAP系列基因,
LOC系列基因,
MIR系列基因,
LINC系列基因
它们往往一个系列,就有好几百个基因;
C12orf44; Chromosome 12 Open Reading Frame 44; 这个是C orf系列基因的意思
MIR系列基因应该是 miRNA相关的基因
LINC系列基因应该就是long intergenic non-protein coding RNA
LOC系列基因,是非正式的,推定的,日后可能被更合适的名字替代
我这里做好了所有的基因对应关系,去生信菜鸟团QQ群里下载吧,共47938个基因的symbol和entrez gene id还有name,还有alias的对应!
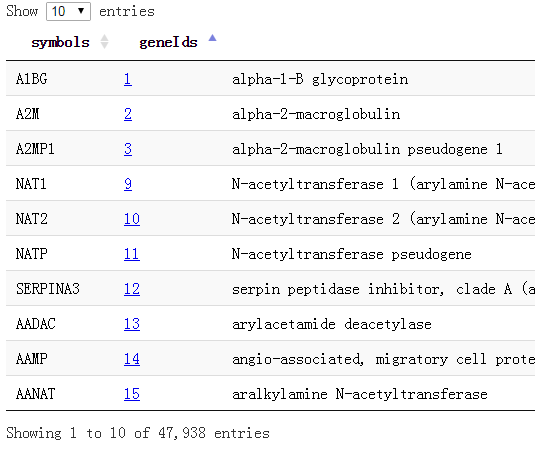
还有一些RNA基因,根本就没有symbol,比如:CTA/B/C/D系列的
Aliases for ENSG00000271971 Gene
Quality Score for this RNA gene is 1
Aliases for ENSG00000271971 Gene
CTD-2006H14.2 5
External Ids for ENSG00000271971 Gene
Ensembl: ENSG00000271971
还有,如果你看到HS.开头的基因,它是unigene的ID了,已经不再是symbol啦。
这是系列文章,请先看:
ncbi现有的GPL已经过万了,但是bioconductor的芯片注释包不到一千,虽然bioconductor可以解决我们大部分的需要,比如affymetrix的95,133系列,深圳1.0st系列,HTA2.0系列,但是如果碰到比较生僻的芯片,bioconductor也不会刻意为之制作一个bioconductor的包,这时候就需要自行下载NCBI的GPL信息了,也可以通过R来解决:
##本质上是下载一个文件,读进R里面,然后解析行列式,得到芯片探针与基因的对应关系,看下面的代码,你就能理解了。 Continue reading

昨天我们说到,测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件。SAM的全称是sequence alignment/map format,而BAM就是SAM的二进制文件。通常sam文件太大,我们会生成bam文件来节省空间。sam文件和bam文件的转换用samtools这个软件就可以完成。 Continue reading
质控之前我们在直播八的时候分析过,公司也给了我质控后的的数据,但是毕竟是别人做的,我们做为一个数据分析师,自己动手来验证一下公司给出的报告也是再好不过的了。大家可以跟着我先将下载数据进行一下质控。 Continue reading
上一次直播中,我们对拿到手的测序数据进行了质控,测序数据的质量已经得到了保证。那么接下来就可以把它拿来与参考基因组比对了,这里我们先用参考基因组hg19,大家可以参照【直播】我的基因组(五):测试数据及参考基因组的准备来下载参考基因组hg19,我这里选择的是UCSC提供的hg19。然后安装bwa软件进行比对,可以参考【直播】我的基因组(四):计算资源的准备来安装,以及对hg19建立索引。 Continue reading
时隔好几个月,因为各种各样的原因数据终于拿到了自己的手上,真是不容易啊!
拿到数据后,第一件要做的事情就是检查数据传输的完整性,然后备份!我拿到的数据如下:
可以看到,公司给了我测序仪的下机数据(raw data)和他们质控后的clean data,这个过程减少了6G的数据量,对应着约90亿bp的碱基,相当于减少了3个人的全基因组数据。具体推算公式见前面的系列直播贴!
首先我把数据拷贝到了我上上周买的2T移动硬盘里面,再拷贝到我工作电脑一份,服务器一份,私人电脑一份,另外一个移动硬盘一份。然后删除了公司寄给我的硬盘里面的数据,再把硬盘寄回给公司,然后监督他们删除我所有的数据。(做这么多就是为了保护隐私,当然这个大前提是我已经确定数据没有问题了。)
检查数据传输的完整性就是md5校验,看看数据在拷贝过程中有没有意外的损坏(这个在之前下载数据的时候我也说过)!一般传输数据之前,会用md5命令来生成各个文件的md5值,就是下面的MD5.txt文件里面的内容,然后传输数据之后,需要自行用md5sum -c MD5.txt 来校验文件里面记录的文件的完整性,如果显示都是OK,说明文件拷贝传输过程是没有问题的!但这个过程会耗费大量的磁盘读写,磁盘读写能力是有限的,所以开多个进程并不能加快这一过程。
然后我把公司处理好的bam文件上传到服务器做下游分析,我用的winscp软件把文件传到服务器上的!
从明天起,我们就开始正式对基因组进行分析啦!欢迎围观!
请扫描以下二维码关注我们,获取直播系列的所有帖子!

首先要明白这个ES score图片里面的数据是什么,这样才能修改它,因为java是一个封闭打包好的软件,所以我们没办法在里面修改它没有提供的参数,运行完GSEA,默认输出的图就是下面这样: Continue reading
GSEA这个java软件使用非常方便,只需要根据要求做好GCT/CLS格式的input文件就好了。我以前也写个用法教程:
mkdir -p ~/annotation/variation/human/dbSNPcd ~/annotation/variation/human/dbSNP## https://www.ncbi.nlm.nih.gov/projects/SNP/## ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b147_GRCh38p2/## ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b147_GRCh37p13/nohup wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b147_GRCh37p13/VCF/All_20160601.vcf.gz &wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b147_GRCh37p13/VCF/All_20160601.vcf.gz.tbi