GSEA这个java软件使用非常方便,只需要根据要求做好GCT/CLS格式的input文件就好了。我以前也写个用法教程:
但说到统计学原理,就有点麻烦了,我试着用自己的思路阐释一下:
假设芯片或者其它测量方法测到了2万个基因,那么这两万个基因在case和control组的差异度量(六种差异度量,默认是signal 2 noise,GSEA官网有提供公式,也可以选择大家熟悉的foldchange)肯定不一样,那么根据它们的差异度量,就可以对它们进行排序,并且Z-score标准化,在下图的最底端展示的就是
那么图中间,就是我们每个gene set里面的基因在所有的2万个排序好基因的位置,如果gene set里面的基因集中在2万个基因的前面部分,就是在case里面富集,如果集中在后面部分,就是在control里面富集着。
而最上面的那个ES score的算法,大概如下:
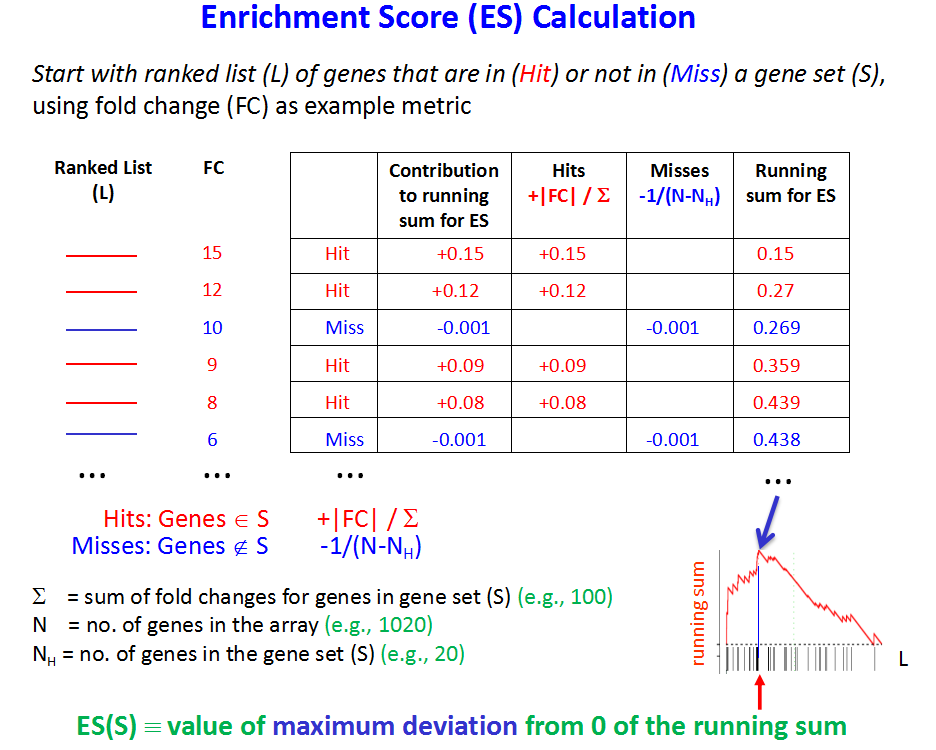
仔细看,其实还是能看明白的,每个基因在每个gene set里面的ES score取决于这个基因是否属于该gene set,还有就是它的差异度量,上图的差异度量就是FC(foldchange),对每个gene set来说,所有的基因的ES score都要一个个加起来,叫做running ES score,在加的过程中,什么时候ES score达到了最大值,就是这个gene set最终的ES score!
算法解读我参考的PPT,反正我是看懂了,但不一定能讲清楚: