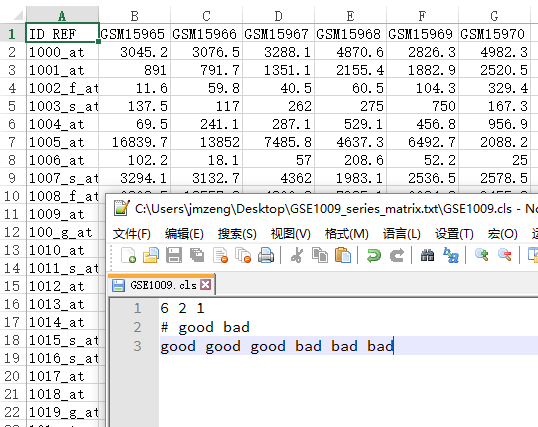
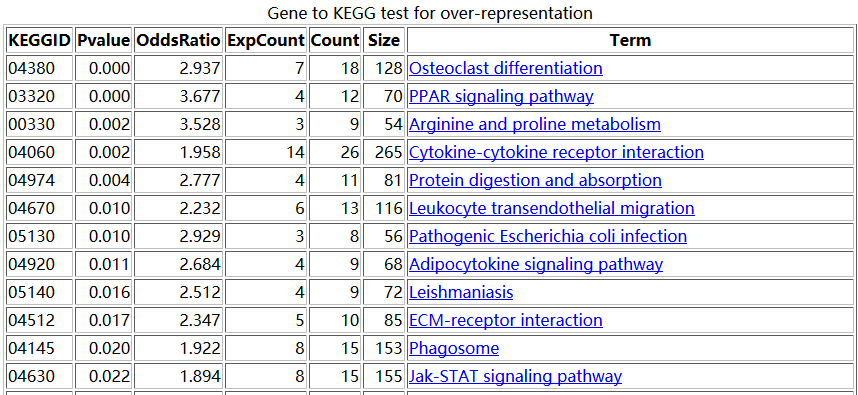
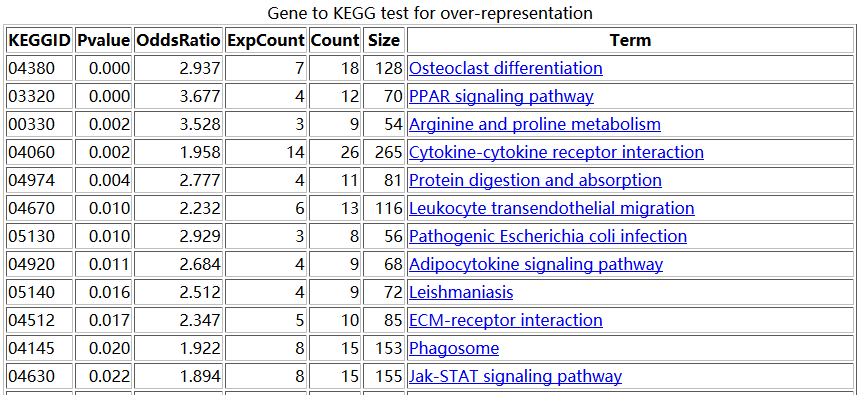
这里的拷贝数变异检测芯片指的是Affymetrix Genome-Wide Human SNP Array 6.0
cel数据,需要处理成segment及genotype数据
这个芯片在TCGA计划里面用的非常多,是标配了。大家只要记住,这是一个跟拷贝数变异检测相关的芯片,而且还可以测一些genotype
Affymetrix Genome-Wide Human SNP Array 6.0是唯一可以真正将CNP(拷贝数多态性)转化成高分辨率的参考图谱的平台。主要应用领域包括全基因组SNP分型、全基因组CNV分型、全基因组关联 分析、全基因组连锁分析。除了进行基因分型外,还为拷贝数研究和LOH研究提供帮助,从而能够进行:UPD检测、亲子鉴定、异常的亲代起源分析(针对 UPD和缺失)、纯合性分析、血缘关系鉴定。
SNP Array 6.0是昂飞公司继Mapping10k、100k、500k和SNP5.0芯片后推出的新一代SNP芯片。在一张芯片上可以分析一个样本906,600 个SNP的基因型, 大约有482,000个SNP来自于前代产品500K和SNP5.0芯片。剩下424,000个SNP包括了来源于国际HapMap计划中的标签 SNP,X,Y染色体和线粒体上更具代表性的SNP,以及来自于重组热点区域和500K芯片设计完成后新加入dbSNP数据库的SNP。该芯片同时含 946,000个非多态性CNV探针,用于检测拷贝数变异,其中202,000个用于检测5677个已知拷贝数变异区域的探针,这些区域来源于多伦多基因 组变异体数据库。该数据库中每隔3,182个非重叠片段区域分别用61个探针来检测。除了检测这些已知的拷贝数多态区域,还有超过744,000个探针平 均分配到整个基因组上,用来发现未知的拷贝数变异区域。SNP和CNV两种探针高密度且均匀地分布在整个基因组,作为拷贝数变异和杂合性缺失(LOH)检 测的工具来发现微小的染色体增加和缺失。为广大生命科学研究者提高发现复杂疾病相关基因的可能提供了强有力的工具。
通过与哈佛大学合办的Broad研究所合作,SNP6.0芯片在数据准确性和一致性方面达到了新的高度。相应推出的Genotyping Console用来处理SNP6.0芯片数据和全基因组遗传分析及质量控制。
通过与哈佛大学合办的Broad研究所合作,SNP6.0芯片在数据准确性和一致性方面达到了新的高度。相应推出的Genotyping Console用来处理SNP6.0芯片数据和全基因组遗传分析及质量控制。
产品特点:
1.涵盖超过1,800,000个遗传变异标志物:包括超过906,600个SNP和超过946,000个用于检测拷贝数变化(CNV,Copy Number Variation)的探针;
2.SNP和CNV两种探针高密度且均匀地分布在整个基因组,不仅可以用于SNP基因精确分型,还可用于拷贝数变异CNV的研究;
3.744,000个探针平均分配到整个基因组上,用来发现未知的拷贝数变异区域;
4.可用于Copy-neutral LOH/UPD检测,亲子鉴定,纯合性分析、血缘关系鉴定、遗传病或其它疾病的研究。
在NCBI的GEO数据库里面可以查到这个芯片,已经有一万多个样本数据啦!
图中第一个是CCLE计划的近千个样本,可能是定制化了的snp6.0芯片吧
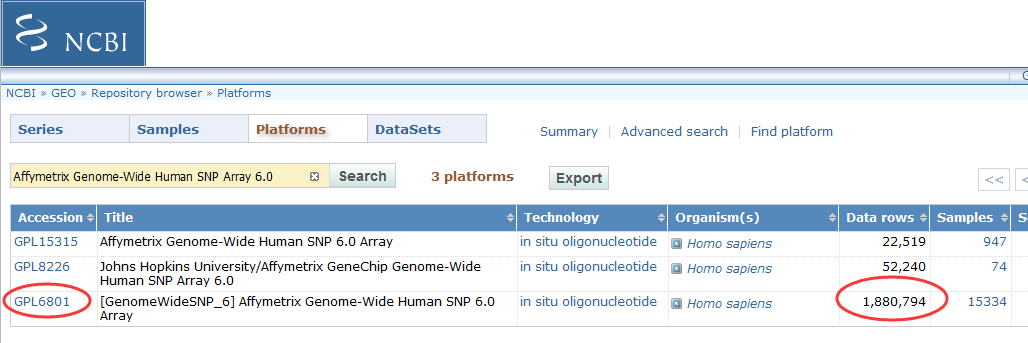
使用这个芯片数据来发文章的非常多,见列表:http://media.affymetrix.com/support/technical/other/snp6_array_publications.pdf
还有一篇2010-nature文章讲了如何用picnic来研究cnv,http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3145113/
也有一篇2010年的文章提出了新的软件来分析这个芯片cnv数据http://bioinformatics.oxfordjournals.org/content/26/11/1395.long
实现同样功能的软件,非常之多,还有一个R的bioconductor系列的包

随便进去都可以找到很多raw data,可以自己进行分析的!