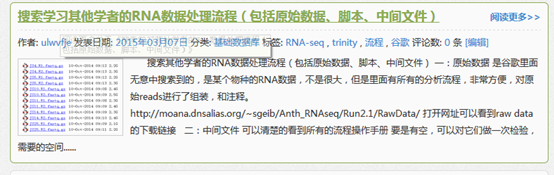
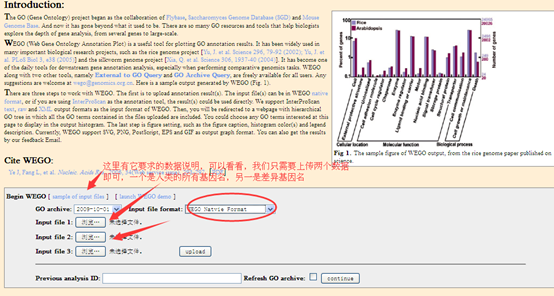
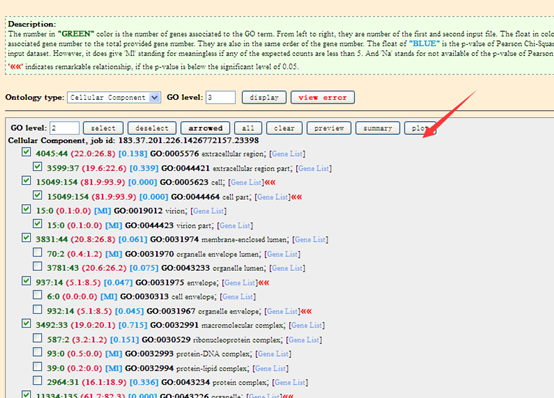
转录组方向:
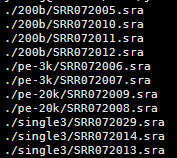
数据来源是NCBI里面的一个文献
其中转录组方向的那些软件流程大多已经跑完了,大家可以见我的转录组总结。
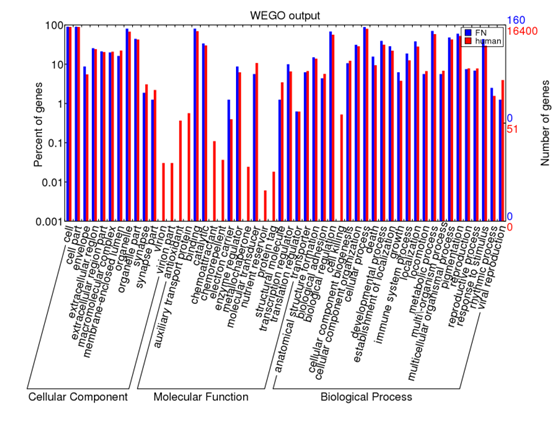
trinity,tophat,cufflinks,RseQC,RNAseq,GOseq,MISO,RSEM,khmer,screed,trimmomatic,transDecoder,vast-tools,picard-tools,htseq,cuffdiff,edgeR,DEseq,funnet,davidgo,wego,kobas,KEGG,Amigo,go
基因组方向:
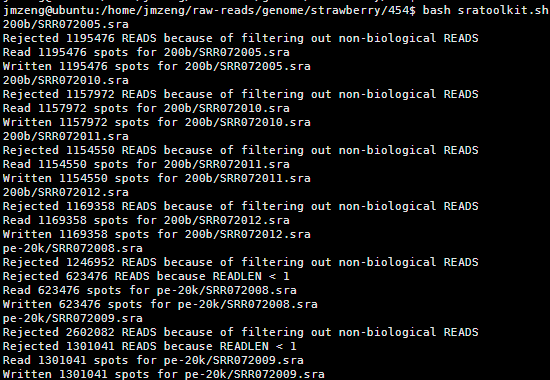
数据来源是strawberry草莓的文献
velvet,SOAPdenovo2,repeatmasker,repeatscount,piler,
Chip-seq方向:
这个群里有高手说要跟我合作,他来帮我写,希望是真的!
免疫组库方向:
这个其实没有成熟软件,也就是一个igblastn, 然后是IMGT数据库,但是是我主打的产品,所以我会详细介绍一下。
全外显子组方向:
![]()
这方面我不是很懂,。好像主要就是snp-calling
Snp-calling方向:
这个我准备自己写软件,不仅仅是用别人的软,它的数据本身也是前面几个方向的数据
bwa,bowtie,samtools,GATK,VarScan.jar,annovar
进化方向:
数据就是基因组数据
orthMCL,inparanoid, clustw,muscle,MAFFT,quickparanoid,blast2go,RAxML,phyML