脚本类似于下面的样子,大家可以读懂之后就仿写
for i in *sra
do
echo $i
/home/jmzeng/bio-soft/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump --split-3 $i
Done
这个脚本是把当前目录下所有的NCBI下载的sra文件都加压开来成测序fastq格式文件
有这些数据,分布在不同的目录,如果是写命令一个个文件处理,很麻烦,如果有几百个那就更麻烦了,所以需要用shell脚本
这样只需要bash这个脚本即可一次性处理所有的数据
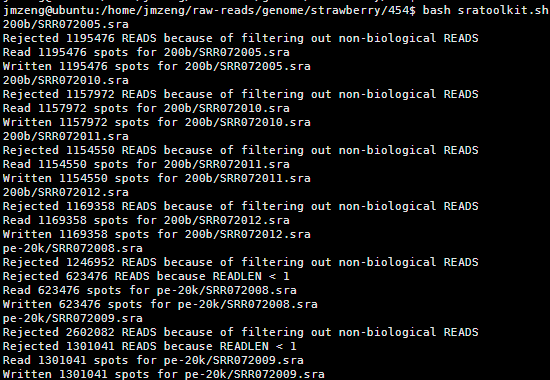
还有很多类似的脚本,非常简单的
for i in *fq
do
echo $i
bowtie2 -p 13 -x ../../RNA.fa -U $i -S $i.sam
done
for i in */accepted_hits.bam
do
echo $i
out=`echo $i |cut -d'/' -f 1`_clout
samtools mpileup -guSDf /home/immune/refer_genome/hg19/hg19.fa $i | bcftools view -cvNg - >snp-vcf/$out.vcf
done
while read id
do
echo $id
out=`echo $id |cut -d'/' -f 2`
reads=`echo $id |cut -d'/' -f 3|sed 's/\r//g'`
tophat2 -p 13 -o $out /home/immune/refer_genome/hg19/hg19 $reads
done <$1
等等