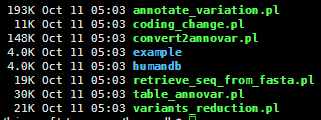
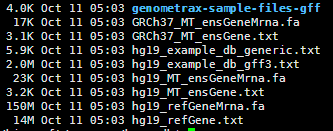
http://statgenpro.psychiatry.hku.hk/limx/kggseq/download/resources/
这个网站收集了大部分资料,我们就用它的,如果它倒闭了,大家再想办法去搜索吧。
其实这些文件都是基于NCBI以及UCSC和ensembl数据库的文件用一些脚本转换而来的,都是非常简单的脚本。
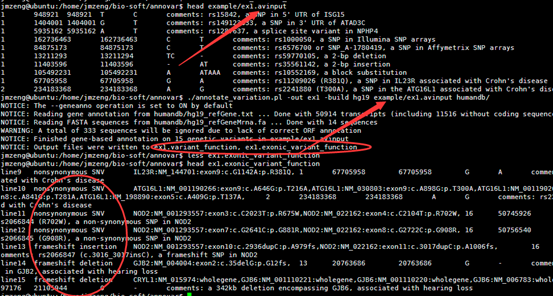
首先我们看看humandb/hg19_refGene.txt 这个文件,总共2.5万多个基因的共5万多个转录本。
19 可能是entrez ID,但是又不像。
NM_001291929 参考基因名
chr11 染色体
-
89057521
89223909
89059923
89223852
17 89057521,89069012,89070614,89073230,89075241,89088129,89106599,89133184,89133382,89135493,89155069,89165951,89173855,89177302,89182607,89184952,89223774, 89060044,89069113,89070683,89073339,89075361,89088211,89106660,89133247,89133547,89135710,89155150,89166024,89173883,89177400,89182692,89185063,89223909,
0
NOX4 基因的英文简称,通俗名
cmpl
cmpl
2,0,0,2,2,1,0,0,0,2,2,1,0,1,0,0,0,
然后我们看看hg19_snp141.txt这个文件
1 10229 A - .
1 10229 AACCCCTAACCCTAACCCTAAACCCTA - .
1 10231 C A .
1 10231 C - .
1 10234 C T .
1 10248 A T .
1 10250 A C .
1 10250 AC - .
1 10255 A - .
1 10257 A C .
1 10259 C A .
1 10291 C T .
1 10327 T C .
1 10329 ACCCCTAACCCTAACCCTAACCCT - .
1 10330 C - .
1 10390 C - .
1 10440 C A .
1 10440 C - .
1 10469 C G .
1 10492 C T .
1 10493 C A .
1 10519 G C .
1 10583 G A 0.144169
1 10603 G A .
1 10611 C G 0.0188246
1 10617 CGCCGTTGCAAAGGCGCGCCG -
里面记录了以hg19为参考的所有的snp位点。
585
ENST00000518655 基因的ensembl ID号
chr1 + 11873 14409 14409 14409
4 基因有四个外显子
11873,12594,13402,13660, 12227,12721,13655,14409, 在基因的四个外显子的坐标
0
DDX11L1 基因的通俗英文名
none none -1,-1,-1,-1,
CTTGCCGTCAGCCTTTTCTTT·····gene的核苷酸序列