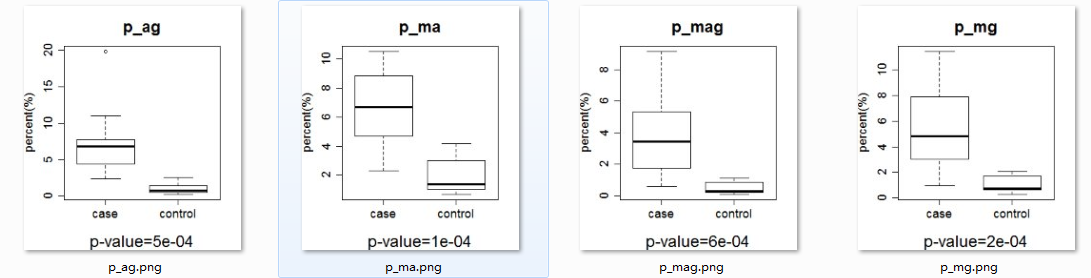
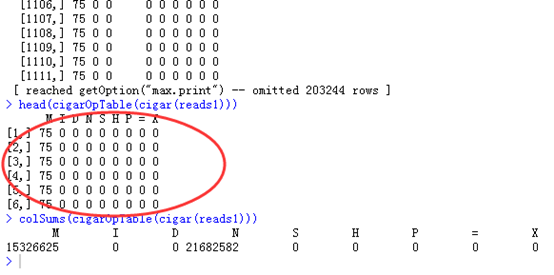
需要做T检验的的数据如下:其中加粗加黑的是case,其余的是control,需要进行检验的是
p_ma p_mg p_ag p_mag 这四列数据,在case和control里面的差异,当然用肉眼很容易看出,control要比case高很多
| individual | m | a | g | ma | mg | ag | mag | p_ma | p_mg | p_ag | p_mag |
| HBV10-1 | 33146 | 3606 | 2208 | 308 | 111 | 97 | 39 | 0.79% | 0.29% | 0.25% | 0.10% |
| HBV15-1 | 23580 | 10300 | 3140 | 1469 | 598 | 560 | 323 | 4.19% | 1.71% | 1.60% | 0.92% |
| HBV3-1 | 107856 | 26445 | 15077 | 4773 | 2383 | 1869 | 1130 | 3.34% | 1.67% | 1.31% | 0.79% |
| HBV4-1 | 32763 | 6448 | 4384 | 579 | 291 | 295 | 124 | 1.35% | 0.68% | 0.69% | 0.29% |
| HBV6-1 | 122396 | 6108 | 7953 | 911 | 796 | 347 | 144 | 0.67% | 0.59% | 0.26% | 0.11% |
| IGA-1 | 31337 | 22167 | 14195 | 3851 | 2752 | 4101 | 2028 | 6.50% | 4.65% | 6.92% | 3.42% |
| IGA-10 | 6713 | 9088 | 12801 | 2275 | 2470 | 4284 | 1977 | 10.54% | 11.44% | 19.85% | 9.16% |
| IGA-11 | 61574 | 23622 | 15076 | 5978 | 4319 | 3908 | 2618 | 6.71% | 4.84% | 4.38% | 2.94% |
| IGA-12 | 38510 | 23353 | 20148 | 6941 | 6201 | 6510 | 4328 | 10.38% | 9.27% | 9.73% | 6.47% |
| IgA-13 | 155379 | 81980 | 65315 | 26055 | 20085 | 17070 | 10043 | 10.38% | 8.00% | 6.80% | 4.00% |
| IgA-14 | 345430 | 86462 | 71099 | 27541 | 21254 | 10923 | 6435 | 6.06% | 4.67% | 2.40% | 1.42% |
| IgA-15 | 3864 | 3076 | 1942 | 378 | 207 | 389 | 145 | 4.66% | 2.55% | 4.80% | 1.79% |
| IgA-16 | 3591 | 4106 | 2358 | 427 | 174 | 424 | 114 | 4.64% | 1.89% | 4.61% | 1.24% |
| IgA-17 | 893 | 1442 | 799 | 68 | 28 | 78 | 18 | 2.27% | 0.94% | 2.61% | 0.60% |
| IGA-2 | 23097 | 5083 | 5689 | 910 | 951 | 1173 | 549 | 2.89% | 3.02% | 3.72% | 1.74% |
| IGA-3 | 14058 | 9364 | 8565 | 2130 | 1953 | 2931 | 1436 | 8.03% | 7.36% | 11.05% | 5.41% |
| IGA-4 | 81571 | 36894 | 33664 | 11346 | 10131 | 9908 | 6851 | 8.86% | 7.91% | 7.74% | 5.35% |
| IGA-5 | 27626 | 6492 | 4503 | 963 | 752 | 942 | 410 | 2.64% | 2.06% | 2.58% | 1.12% |
| IGA-7 | 55348 | 5956 | 4028 | 833 | 476 | 468 | 207 | 1.30% | 0.74% | 0.73% | 0.32% |
| IGA-8 | 31671 | 17097 | 10443 | 3894 | 2666 | 3514 | 2003 | 7.56% | 5.17% | 6.82% | 3.89% |
但是我们需要写程序对这些百分比在case和control里面进行T检验。
a=read.table("venn_dat.txt",header=T)
type=c(rep("control",5),rep("case",12),"control","control","case")
for (i in 9:12)
{
dat=as.numeric(unlist(lapply(a[i],function(x) strsplit(as.character(x),"%"))))
filename=paste(names(a)[i],".png",sep="")
png(file=filename, width = 320, height = 360)
b=t.test(dat~type)
txt=paste("p-value=",round(b$p.value[1],digits=4),sep="")
plot(as.factor(type),dat,ylab="percent(%)",main=names(a)[i],sub=txt,cex.axis=1.5,cex.sub=2,cex.main=2,cex.lab=1.5)
dev.off()
}
得到的图片如下: