如果只是画网络图,那么只需要把所有的点,按照算好的坐标画出来,然后把所有的连线也画出即可!
其中算法就是,点的坐标该如何确定?Two of the most prominente algorithms (Fruchterman & Reingold’s force-directed placement algorithm and Kamada-Kawai’s)
有一个networkD3的包可以直接画图,但是跳过了确定点的坐标这个步骤,我重新找了一包,可以做到!
作者只是用sna包来得到数据,其实用的是ggplot来画网络图!
需要熟悉network包里面的network对象的具体东西,如何自己构造一个,然后数学sna包如何计算layout即可
解读这个包,也可以自己画网络图,代码如下:
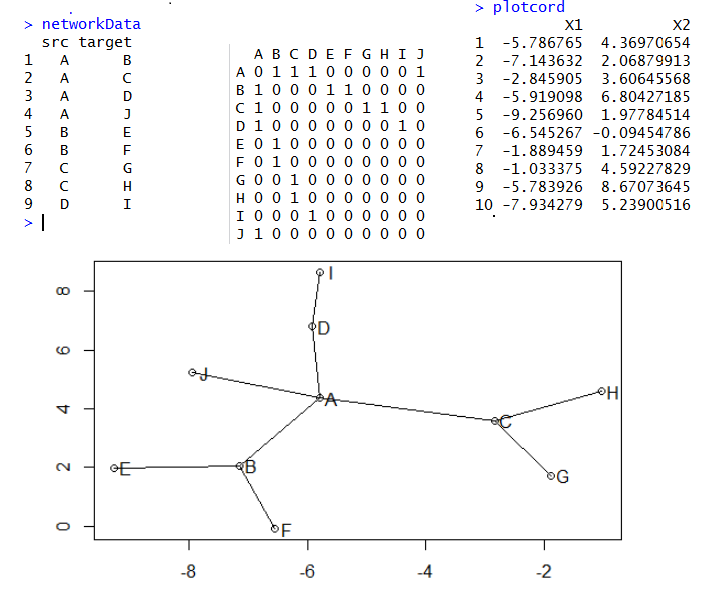
plot(plotcord)
text(x=plotcord$X1+0.2,y=plotcord$X2,labels = LETTERS[1:10])
for (i in 1:10){
for (j in 1:10){
if(tmp[i,j]) lines(plotcord[c language="(i,j),1"][/c],plotcord[c language="(i,j),2"][/c])
}
}
当然,我们还没有涉及到算法,就是如何生成plotcord这个坐标矩阵的!
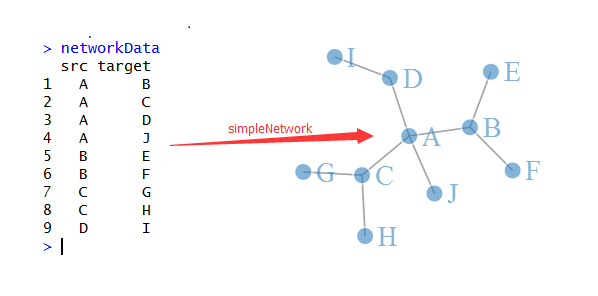
大家看下面这个示意图就知道网络图是怎么样画出来的了,首先我们有一些点,它们之间有联系,都存储在networData这个数据里面,是10个点,共9个连接,然后我用reshape包把它转换成连接矩阵,理论上10个点的两两相互作用应该有100条线,但是我们的数据清楚的说明只有9条,所以只有9个1,其余的0代表点之间没有关系。接下来我们用sna这个包对这个连接矩阵生成了这10个点的坐标(这个是重点),最后很简单了,把点和线画出来即可!
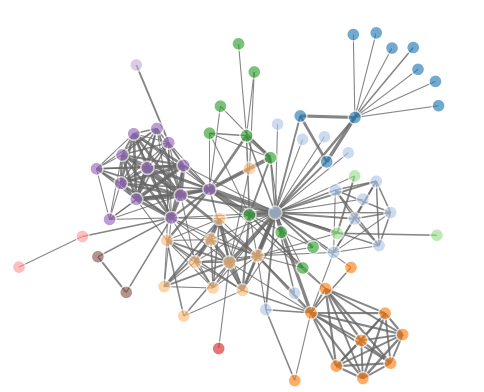
另外一个例子:
net=network(150, directed=FALSE, density=0.03)
m <- as.matrix.network.adjacency(net) # get sociomatrix
# get coordinates from Fruchterman and Reingold's force-directed placement algorithm.
plotcord <- data.frame(gplot.layout.fruchtermanreingold(m, NULL))
# or get it them from Kamada-Kawai's algorithm:
# plotcord <- data.frame(gplot.layout.kamadakawai(m, NULL))
colnames(plotcord) = c("X1","X2") ###所有点的坐标,共150个点
edglist <- as.matrix.network.edgelist(net) ##所有点之间的关系-edge ##共335条线
edges <- data.frame(plotcord[edglist[,1],], plotcord[edglist[,2],]) ##两点之间的连线的具体坐标,335条线的起始终止点点坐标
原始代码如下:
library(network)
library(ggplot2)
library(sna)
library(ergm)

大家可以试用这个代码,因为它用的ggplot,肯定比我那个简单R作图要好看多了
参考算法文献: