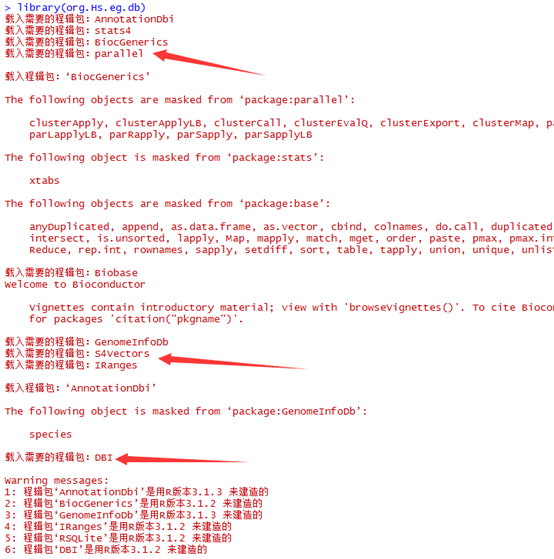
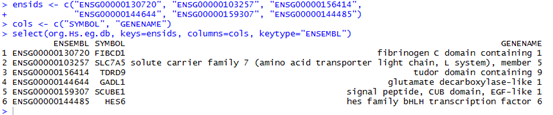
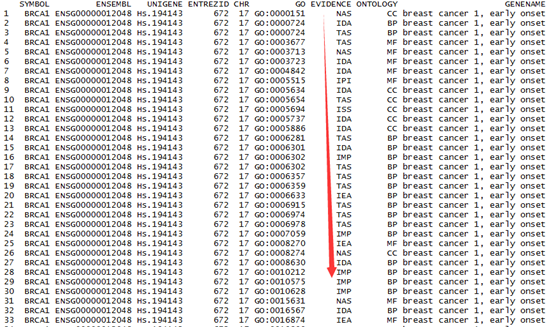
这是发布在bioconductor平台上面的一个数据库文件,可以通过R里面下载安装并使用,非常方便。其实在ensembl数据库里面也有一个biomart,我之前也讲过这个平台,非常好用,可以把任意的数据库之间的ID号进行转换。
为了更好的理解和掌握biomaRt,我们可以先通过在线资源来了解一下它的原型biomart (http://www.biomart.org)。 biomart是为生物科研提供数据服务的免费软件,它为数据下载提供打包方案。它有许多成功的应用实例,比如欧洲生物信息学中心(The European Bioinformatics Institute ,EBI)维护的Ensembl数据库(http://www.ensembl.org/)就使用biomart提供数据批量下载服务, 还有COSMIC, Uniprot, HGNC, Gramene, Wormbase以及dbSNP等。
这个就是一个R平台的biomart而已,但是非常好用!
> library(biomaRt)
> head(listMarts(), 3)
biomart version
1 ensembl ENSEMBL GENES 79 (SANGER UK)
2 snp ENSEMBL VARIATION 79 (SANGER UK)
3 regulation ENSEMBL REGULATION 79 (SANGER UK)
这是这个biomart最具有代表性的三个数据库,用listMarts()可以查看得知,它总共有58个数据库。
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
这是创建了人的ensembl数据库对象
> head(listFilters(ensembl), 3)
name description
1 chromosome_name Chromosome name
2 start Gene Start (bp)
3 end Gene End (bp)
可以看到对人的数据库ensembl来说,有多种字段可以来挑选自己感兴趣的东西,最常用的的当然是染色体号及起始终止坐标啦,用listFilters(ensembl),以查看得知,它总共有284中挑选感兴趣数据的方式。
既然 chromosome_name是其中一个挑选字段,那么我们就可以看看,是如何进行挑选的
用filterOptions(myFilter, ensembl)可以看到它挑选参数非常之多,远不止我们所认为的染色体号码。
染色体号一般是1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,X,Y,MT
还有一堆稀奇古怪的标志,LRG_101,LRG_102,LRG_103,LRG_104,因为我们组装好的人的标准基因组还有很多小的片段不被计入染色体中。
然后还可以看到人的ensembl数据库对象,有很多的属性,最常见的当然是基因ID和转录本ID和蛋白的ID号啦!
> head(listAttributes(ensembl), 3)
name description
1 ensembl_gene_id Ensembl Gene ID
2 ensembl_transcript_id Ensembl Transcript ID
3 ensembl_peptide_id Ensembl Protein ID
用listAttributes(ensembl),,以查看得知,它总共有1166个ID号,太恐怖了,我实在是没有想到!
那么接下来我简单讲讲这个包的几个应用吧
首先是根据entrez ID号来找
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
这样就得到了人的信息,然后我探究以下两个基因的其它信息。
entrzID=c("672","1")
getBM(attributes=c("entrezgene","hgnc_symbol","ensembl_gene_id"), filters = "entrezgene", values =entrzID, mart=ensembl )
entrezgene hgnc_symbol ensembl_gene_id
1 1 A1BG ENSG00000121410
2 672 BRCA1 ENSG00000012048
3 672 BRCA1 LRG_292
其实这个函数很简单,就是根据自定义的entrzID这个变量来找到一些数据,数据的属性是我自己定义的entrezgene","hgnc_symbol","ensembl_gene_id",所以它就显示这个信息给我,在我之前弄好的人的数据库里面寻找!listAttributes(ensembl),,以查看得知,它总共有1166个ID号,就是说,你可以挑选你想要的基因的1166种信息,包罗万象!!!
其它功能也是很简单的啦,自己多看帮助文档!
从上面的操作来看,使用biomaRt只需要两步,1,指定mart数据库,2,使用getBM获得注释。但是首先,我们如何知道有哪些服务器,以及这些服务器上哪些数据库呢?其次,我们如何获阳getBM中attributes,filters的正确设置呢?
关于第一个问题,我们可以使用biomaRt中的listMarts以及listDatasets两个函数来解决。
> marts <- listMarts(); head(marts) #查看当前可用的数据源 ,总共有58个数据源。
> ensembl <- useMart("ensembl") #使用ensembl数据源
> datasets <- listDatasets(ensembl); datasets[1:10,] #查看ensembl中可用数据库,共有69个物种的数据库!
对于第二个问题,我们使用biomaRt中的listFilters以及listAttributes两个函数来解决。
> mart <- useMart("ensembl", "hsapiens_gene_ensembl") #首先使用人的数据库
>listAttributes(ensembl) #,以查看得知,它总共有1166个ID号,就是说,人的数据库可供挑选的信息多达1166种。
> filters <- listFilters(mart); filters[grepl("entrez", filters[,1]),] #总共有284中挑选感兴趣数据的方式。
最后的问题是,biomaRt会被如何使用呢?我们做注释的时候,怎么就想到要使用biomaRt呢?因为在注释上,各种ID,symbol, name之间的转换都可以考虑使用biomaRt来做。更重要的是,biomaRt还会有很多SNP, alternative splicing, exon, intron, 5’utr, 3’utr等等信息。当然,只要能做也数据库并使用SQL访问的数据都可以使用biomaRt来获取。所以我们的思路可以更加发散一些。