做snp-calling时很多标准流程都会提到去除PCR重复这个步骤,但是这个步骤对找snp的影响到底有多大呢?这里我们来探究一下
| 去除PCR重复前 |
样本名 |
去除PCR重复后 |
| 106082 |
BC1-1.snp |
103829 |
| 101443 |
BC1-2.snp |
99500 |
| 103937 |
BC2-1.snp |
101833 |
| 102979 |
BC2-2.snp |
101022 |
| 105876 |
BC3-1.snp |
103562 |
| 109168 |
BC3-2.snp |
107052 |
| 107155 |
BC4-1.snp |
104894 |
| 108335 |
BC4-2.snp |
106031 |
| 100236 |
BC5-1.snp |
98417 |
| 102322 |
BC5-2.snp |
100395 |
| 103466 |
BC6-1.snp |
101405 |
| 112940 |
BC6-2.snp |
110611 |
| 113166 |
BC7-1.snp |
110948 |
| 114038 |
BC7-2.snp |
116090 |
| 123670 |
PC1-1.snp |
121697 |
| 111402 |
PC1-2.snp |
109389 |
| 106917 |
PC2-1.snp |
105149 |
| 108724 |
PC2-2.snp |
106776 |
可以看到去除pcr重复这个脚本对snp-calling的结果影响甚小,就是少了那么一千多个snp,脚本如下,我是用picard-tools进行的去除PCR重复,当然也可以用samtools来进行同样的步骤
[shell]
<b>for i in *.sorted.bam</b>
<b>do</b>
<b>echo $i</b>
<b>java -Xmx120g -jar /home/jmzeng/snp-calling/resources/apps/picard-tools-1.119/MarkDuplicates.jar \</b>
<b>CREATE_INDEX=true REMOVE_DUPLICATES=True \</b>
<b>ASSUME_SORTED=True VALIDATION_STRINGENCY=LENIENT METRICS_FILE=/dev/null \</b>
<b>INPUT=$i OUTPUT=${i%%.*}.sort.dedup.bam</b>
<b>done</b>
[/shell]
然后我们首先看看没有产生变化的那些snp信息的改变
head -50 ../rmdup/out/snp/BC1-1.snp |tail |cut -f 1,2,8
chr1 17222 ADP=428;WT=0;HET=1;HOM=0;NC=0
chr1 17999 ADP=185;WT=0;HET=1;HOM=0;NC=0
chr1 18091 ADP=147;WT=0;HET=1;HOM=0;NC=0
chr1 18200 ADP=278;WT=0;HET=1;HOM=0;NC=0
chr1 24786 ADP=238;WT=0;HET=1;HOM=0;NC=0
chr1 25072 ADP=24;WT=0;HET=1;HOM=0;NC=0
chr1 29256 ADP=44;WT=0;HET=1;HOM=0;NC=0
chr1 29265 ADP=44;WT=0;HET=1;HOM=0;NC=0
chr1 29790 ADP=351;WT=0;HET=1;HOM=0;NC=0
chr1 29939 ADP=109;WT=0;HET=1;HOM=0;NC=0
head -50 BC1-1.snp |tail |cut -f 1,2,8
chr1 17222 ADP=457;WT=0;HET=1;HOM=0;NC=0
chr1 17999 ADP=196;WT=0;HET=1;HOM=0;NC=0
chr1 18091 ADP=155;WT=0;HET=1;HOM=0;NC=0
chr1 18200 ADP=313;WT=0;HET=1;HOM=0;NC=0
chr1 24786 ADP=254;WT=0;HET=1;HOM=0;NC=0
chr1 25072 ADP=25;WT=0;HET=1;HOM=0;NC=0
chr1 29256 ADP=46;WT=0;HET=1;HOM=0;NC=0
chr1 29265 ADP=46;WT=0;HET=1;HOM=0;NC=0
chr1 29790 ADP=440;WT=0;HET=1;HOM=0;NC=0
chr1 29939 ADP=123;WT=0;HET=1;HOM=0;NC=0
可以看到,同一位点的snp仍然可以找到,仅仅是对测序深度产生了影响
然后我们再看看去除PCR重复这个步骤减少了的snp,在原snp里面是怎么样的
perl -alne '{$file++ if eof(ARGV);unless ($file){$hash{"$F[0]_$F[1]"}=1} else {print if not exists $hash{"$F[0]_$F[1]"} } }' ../rmdup/out/snp/BC1-1.snp BC1-1.snp |less
这个脚本就可以把去除PCR重复找到的snp位点在没有去除PCR重复的找到的snp文件里面过滤掉,查看那些去除PCR重复之前独有的snp
Min. 1st Qu. Median Mean 3rd Qu. Max.
8.00 8.00 11.00 44.26 25.00 7966.00
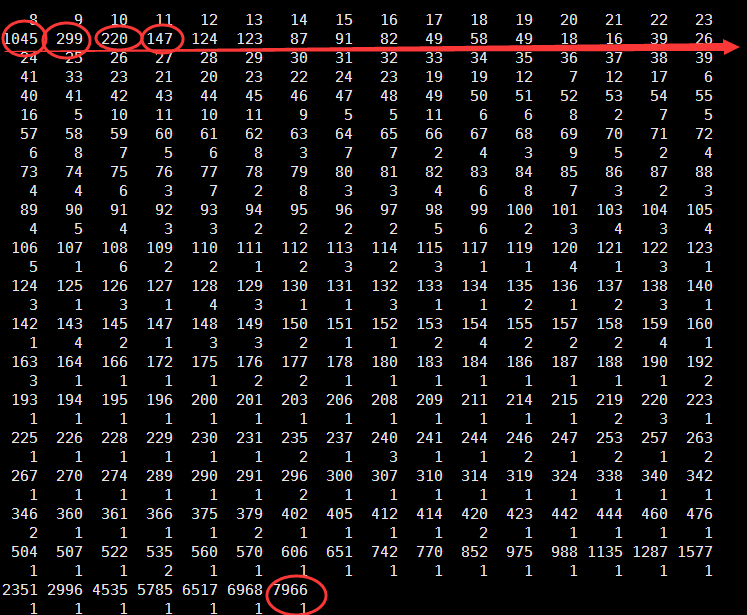
可以看到被过滤的snp大多都是测序深度太低了的,如下面的例子
chr1 726325 a 9 CCC.ccc,^:, IEHGHHG/9
chr1 726325 a 5 C.c,^:, IGH/9
chr1 726338 g 16 TTT.ttt,,....,,, IHGI:9<HIIFIHC5H
chr1 726338 g 10 T.t,,...,, II:HIIFH5H
可以看到这一步还是很有用的,但是怎么说呢,因为最后对snp的过滤本来就包含了一个步骤是对snp的测序深度小于20的给过滤掉
但是也有个别的测序深度非常高的snp居然也是被去除PCR重复这个步骤给搞没了!很奇怪,我还在探索之中.
grep 13777 BC1-1.mpileup |head
chr1 13777 G 263 ........,.C,,,,,.,,,.......,,,..,....,,......,.....c,........,,,,,,,..,...,,,,,.........,......C.......,,,,,,,,,,.....,,,,,,,.,,,..C,,,,,,CC,c,,,...C..,,,,cC.C..CC.CC,,cc,.C...C,,,,CCc,c,,,,,,,c,C.C.CC...C.cc,c...,C.CCcc...,CCC.C.CC..CCC..CC.c,cc,cc,,cc,C.,,^!.^6.^6.^6.^!, HIHIIIIEIEIHGIIIFIHIG?IIIIHIIHIFHIIHICIIIHIIGIEIIGIIIGHIIIIIIHIIHIHIIIIIIIHII1I?GHHHEHHIIEIEHIIEIIHHIIFIIIFHIHIIIIHIHIIHIIHHIIEIIIIIIHIIIIIIIIIG1HIIIIHIHIEHIHIHIIIIIIIIIIIHICIHIIIIIEIIIIHICIHGGIIIIIIIIEHIHIIIIIIHFIGGIIIIGIIIGICIIIHIIIIIIIIIIIHHHIIIIIHIIHDDII>>>>>
grep 13777 BC1-1.rmdup.mpileup |head
chr1 13777 G 240 ........,.C,,,,,.,,,.......,,,..,....,,......,....c,......,,,,,,,..,...,,,,,.........,......C......,,,,,,,,,,.....,,,,,,,.,,,..C,,,,,,CC,c,,,...C..,,,,cC..CC.CC,cc,.C...C,,,,CCc,c,,,,,,,cC.C.C..C.c,c...,C.CCcc...,CC.C.CCC..C.c,cc,,c,.,,^!.^6.^6.^!, HIHIIIIEIEIHGIIIFIHIG?IIIIHIIHIFHIIHICIIIHIIGIEIIIIIHIIIIIHIIHIHIIIIIIIHII1I?GHHHEHHIIEIEHIIEIHHIIFIIIFHIHIIIIHIHIIHIIHHIIEIIIIIIHIIIIIIIIIG1HIIIIHIHIEHIHIIIIIIIIIIHICIHIIIIIEIIIIHICIHGGIIIIIIHIHIIIIIHFIGGIIIIGIIIGCIIIIIIIIIIHHIIIHIHDII>>>>
然后我再搜索了一些
chr8 43092928 . A T . PASS ADP=7966;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:7967:7966:6261:1663:20.9%:0E0:39:39:3647:2614:1224:439
chr8 43092908 . T C . PASS ADP=6968;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:7002:6968:5315:1537:22.06%:0E0:37:38:3022:2293:890:647
chr8 43092898 . T G . PASS ADP=6517;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:6517:6517:4580:1587:24.35%:0E0:38:38:2533:2047:920:667
chr7 100642950 . T C . PASS ADP=770;WT=0;HET=1;HOM=0;NC=0 GT:GQ:SDP:DP:RD:AD:FREQ:PVAL:RBQ:ABQ:RDF:RDR:ADF:ADR 0/1:255:771:770:615:155:20.13%:3.9035E-51:38:38:277:338:65:90
终于发现规律啦!!!原来它们的突变率都略高于20%,在没有去处PCR重复之前,是高于snp的阈值的,但是去除PCR重复对该位点的突变率产生了影响,使之未能通过筛选。