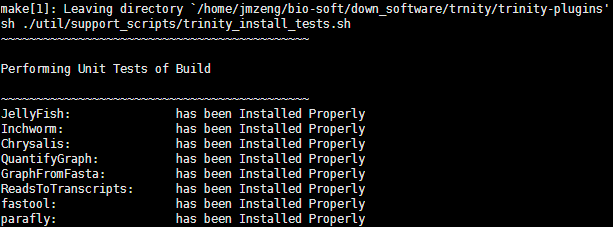
网站成立也快一个月了,总算是完全搞定了生信领域的一个方向,当然,只是在菜鸟层面上的搞定,还有很多深层次的应用及挖掘,仅仅是我所讲解的这些软件也有多如羊毛的参数可以变幻,复杂的很。其实我最擅长的并不是转录组,但是因为一些特殊的原因,我恰好做了三个转录组项目,所以手头上关于它的资料比较多,就分享给大家啦!稍后我会列一个网站更新计划,就好谈到我所擅长的基因组及免疫组库。我这里简单对转录组做一个总结:
首先当然是我的转录组分类网站啦
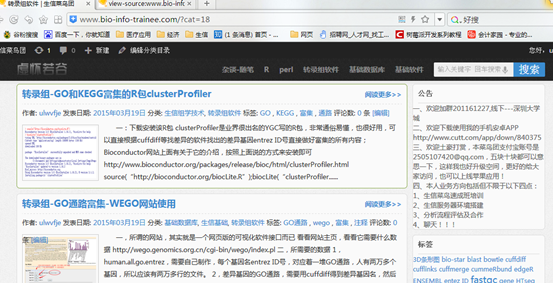
http://www.bio-info-trainee.com/?cat=18
同样的我用脚本总结一下给大家
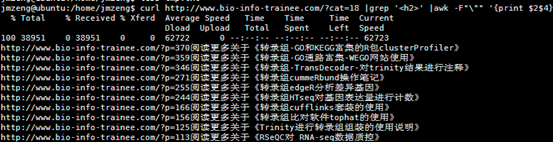
http://www.bio-info-trainee.com/?p=370阅读更多关于《转录组-GO和KEGG富集的R包clusterProfiler》
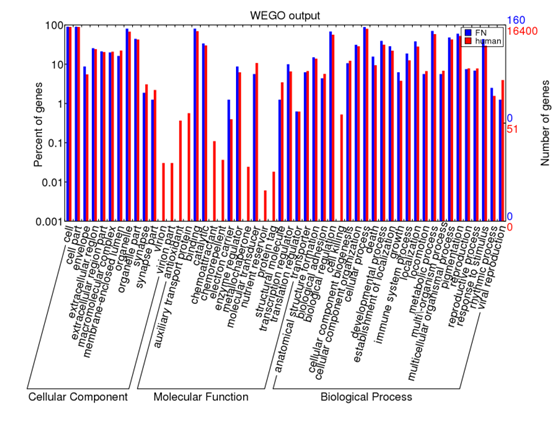
http://www.bio-info-trainee.com/?p=359阅读更多关于《转录组-GO通路富集-WEGO网站使用》
http://www.bio-info-trainee.com/?p=346阅读更多关于《转录组-TransDecoder-对trinity结果进行注释》
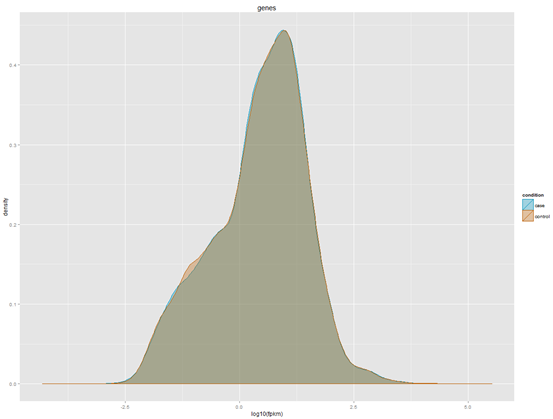
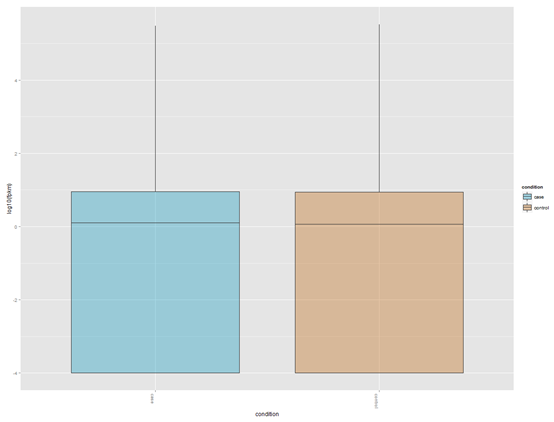
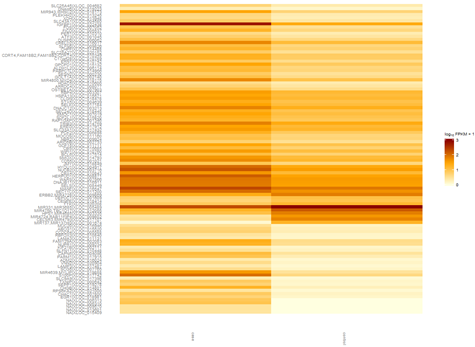
http://www.bio-info-trainee.com/?p=271阅读更多关于《转录组cummeRbund操作笔记》
http://www.bio-info-trainee.com/?p=255阅读更多关于《转录组edgeR分析差异基因》
http://www.bio-info-trainee.com/?p=244阅读更多关于《转录组HTseq对基因表达量进行计数》
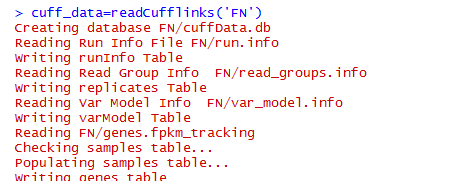
http://www.bio-info-trainee.com/?p=166阅读更多关于《转录组cufflinks套装的使用》
http://www.bio-info-trainee.com/?p=156阅读更多关于《转录组比对软件tophat的使用》
http://www.bio-info-trainee.com/?p=125阅读更多关于《Trinity进行转录组组装的使用说明》
http://www.bio-info-trainee.com/?p=113阅读更多关于《RSeQC对 RNA-seq数据质控》
同时我也讲了如何下载数据
http://www.bio-info-trainee.com/?p=32
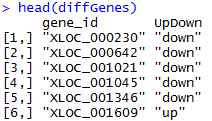
原始SRA数据首先用SRAtoolkit数据解压,然后进行过滤,评估质量,然后trinity组装,然后对组装好的进行注释,然后走另一条路进行差异基因,差异基因有tophat+cufflinks+cummeRbund,也有HTseq 和edgeR等等,然后是GO和KEGG通路注释,等等。
在我的群里面共享了所有的代码及帖子内容,欢迎加群201161227,生信菜鸟团!
http://www.bio-info-trainee.com/?p=1
线下交流-生物信息学
同时欢迎下载使用我的手机安卓APP
http://www.cutt.com/app/down/840375