请不要直接拷贝我的代码,需要自己理解,然后打出来,思考我为什么这样写代码。
软件请用最新版,尤其是samtools等被我存储在系统环境变量的,考虑到读者众多,一般的软件我都会自带版本信息的!
我用两个小时,不代表你是两个小时就学会,有些朋友反映学了两个星期才 学会,这很正常,没毛病,不要异想天开两个小时就达到我的水平。
转录组如果只看表达量真的是超级简单,真是超级简单,而且人家作者本来就测是SE50,这种破数据,也就是看表达量用的!
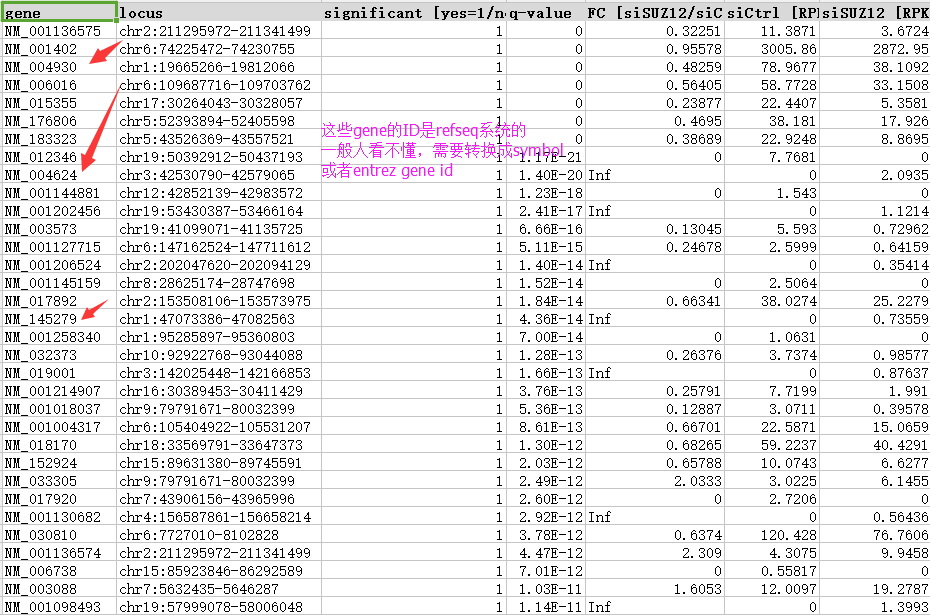
首先作者分析结果是:

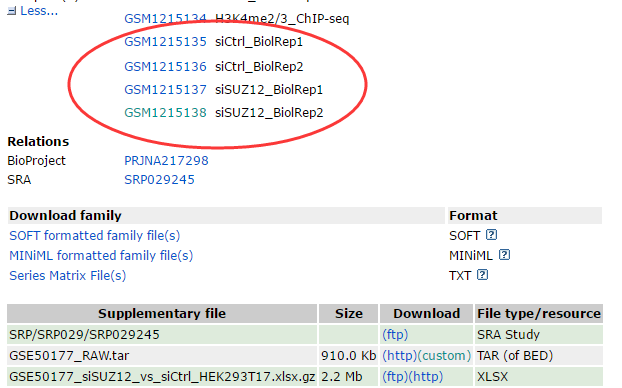
我们需要下载的RNA-seq的数据:

下载地址很容易获取啦!
for ((i=677;i<=680;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP029/SRP029245/SRR957$i/SRR957$i.sra;done
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 $id;done
因为我用fastqc看了看数据质量,发现没有什么问题,代码如下:
ls *fastq |xargs ~/biosoft/fastqc/FastQC/fastqc -t 10
所以直接用hisat2软件把测序得到的fastq文件比对到hg19参考基因组上面
reference=/home/jianmingzeng/reference/index/hisat/hg19/genome
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR957677.fastq -S control_1.sam 2>control_1.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR957678.fastq -S control_2.sam 2>control_2.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR957679.fastq -S siSUZ12_1.sam 2>siSUZ12_1.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR957680.fastq -S siSUZ12_2.sam 2>siSUZ12_2.log
而且查看log日志可以发现,比对效果杠杠的:
93.10% overall alignment rate
92.44% overall alignment rate
92.36% overall alignment rate
93.22% overall alignment rate
92.44% overall alignment rate
92.36% overall alignment rate
93.22% overall alignment rate
然后把sam文件根据reads name来排序并且转换为bam文件节省空间
ls *sam |while read id;do (nohup samtools sort -n -@ 5 -o ${id%%.*}.Nsort.bam $id &);done
最后用htseq-counts工具来对每一个样本进行基因的表达量定量!
ls *.Nsort.bam |while read id;do (nohup samtools view $id | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>${id%%.*}.geneCounts 2>${id%%.*}.HTseq.log&);done
得到的文件如下:
这4个样本的基因的counts数据就可以用一系列的R包来做差异分析了,包括limma的voom,DEseq2,edgeR等等。这些包的用法都烂大街了,我就不赘述了。
做完差异分析,就可以跟作者的结果做对比,看看自己做的是不是对的。