首先载入这个包
source("http://bioconductor.org/biocLite.R")
biocLite("topGO")
biocLite("ALL")
library(topGO)
library(ALL)
data(ALL)
data(geneList)
data(GOdatat)
这样就载入了很多变量, ls()查看如下
[1] "affyLib" "ALL" "geneList" "topDiffGenes"
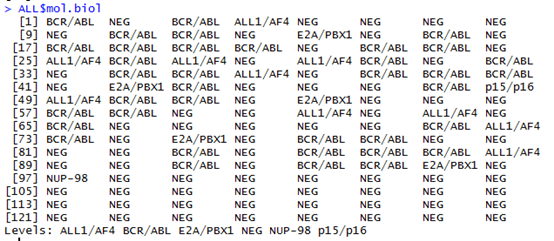
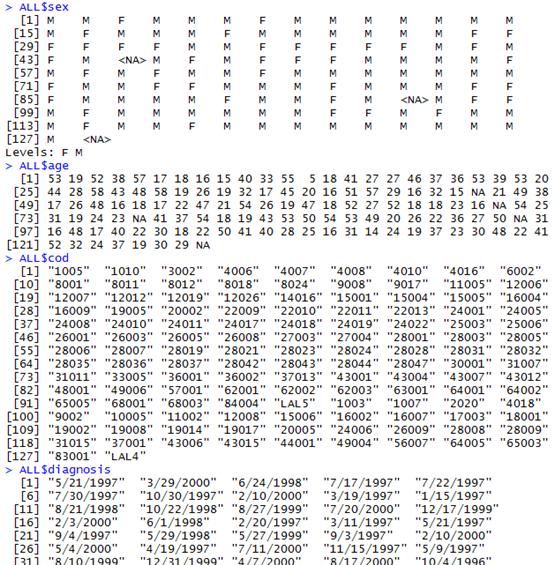
其中ALL这个数据我在另一篇日志里面重点介绍了一下。
然后我简单提一下"geneList"
head(geneList)
1095_s_at 1130_at 1196_at 1329_s_at 1340_s_at 1342_g_at
1.0000000 1.0000000 0.6223795 0.5412240 1.0000000 1.0000000
str(geneList) 是一个向量,有323个数字。
Named num [1:323] 1 1 0.622 0.541 1 ...
- attr(*, "names")= chr [1:323] "1095_s_at" "1130_at" "1196_at" "1329_s_at" ...
然后简单查询该包的安装地址和一些文件
system.file(package = "topGO")
[1] "C:/Program Files/R/R-3.1.1/library/topGO"
在这个目录下面可以找到文件"examples/geneid2go.map"
里面的内容格式如下,第一列是gene的ID号,一般是entrez ID ,第二列是该基因所对应的GO所有的条目,用逗号分隔。
068724 GO:0005488, GO:0003774, GO:0001539, GO:0006935, GO:0009288
119608 GO:0005634, GO:0030528, GO:0006355, GO:0045449, GO:0003677, GO:0007275
此处省略一万行。
readMappings(file = system.file("examples/geneid2go.map", package = "topGO"))
这个函数可以读取我们的文件,返回一个list。是gene到go的映射,每个基因都有一个或者多个go条目。
这个list可以用inverseList这个函数反转,变成每个go条目到基因的映射。
构建topGO这个大全,需要的数据包括:
- 基因identifier,可以附上某种分数以便后面施用某种统计处理,分数可以是t检验的p值或者与某个表型的correlation等;
- identifier和GO term间的map,如果是芯片数据的话BioC里有多种注释包,声明包的名称即可。至于我等蛋白界苦人,也能自己构建map,见下;
- GO的层级结构,由GO.db提供,目前这个包只支持GO.db提供的结构:GOslim就再说了。
topGOdata对象构建函数的参数包括:
- ontology,可指定要分析的GO term的类型,即BP、CC之类;
- description:topGOdata的描述,可选;
- allGenes:基因identifier的原始列表,和后面的geneSelectionFun联合作用,得出参与分析的基因,可以是numeric或factor。
- geneSelectionFun:基因选择函数,如果前面allGenes是numeric的话就必须得指明此参数;
- nodeSize:被认为富集的GO term辖下基因的最小数目(>=),默认为1。
- annotationFun:基因identifier map到GO term的函数。
代码如下
BPterms <- ls(GOBPTerm)
geneID2GO=readMappings(file = system.file("examples/geneid2go.map", package = "topGO"))
直接使用系统自带的data(GOdata)数据,自己构建太麻烦了!
其实就是这就对ALL这个数据集来进行分析!!!
GOdata包含实例topGOdata对象。它可以用来直接运行富集分析。
topGOdata对象构建好后,即可利用这个包里的各种方法和函数做分析。
numGenes(GOdata) 查看对象包含的基因的数目
[1] 318
> description(GOdata)
[1] "Simple topGOdata object"
genes(GOdata)可以得到该对象里面所有的318个基因
geneScore() 可以得到想318个基因的分数
函数sigGenes()返回一个character vector,为各显著变化基因identifier。函数numSigGenes()则用于查看显著变化基因的数目。
函数updateGenes()可以修改topGOdata对象里所包含的基因。
想要看全部基因都对应上了哪些GO term,可用函数usedGO()得到一个character
基因集富集分析(gene set enrichment analysis)
首先看看GSEA的三种方式:
- 基于count,即仅要求输入一组基因,此种方式最为流行,可用Fisher's exact test、Hypegeometric test和binomial test进行检验;
- 基于基因的score或rank,可用Kolmogorov-Smirnov like tests(即05年那篇PNAS的GSEA文章所用方法),Gentleman's Category、t-test等方法;
- 基于基因的表达,可从expression matrix直接分析,如Goeman's globaltest,以及GlobalAncova。
topGO提供两种分析方法,一种自由度更高但上手不易,本菜鸟还是跟着第二种走吧,较为用户友好但集成度较高。简单来说,就是用runTest()这个函数,要求三个主要的argument,一个是之前构建好的topGOdata类实例,第二个参数algorithm用于指定生成GO graph的方法,而参数statistic用于指定所用的检验方法,比如:
> resultFis <- runTest(GOdata, algorithm = "classic", statistic = "fisher")
> resultWeight <- runTest(GOdata, algorithm = "weight", statistic = "fisher")
> resultKS <- runTest(GOdata, algorithm = "classic", statistic = "ks")
> resultKS.elim <- runTest(GOdata, algorithm = "elim", statistic = "ks.elim")
runTest这一锤子买卖敲定后就能开始解读和展示结果了,得到的结果是topGOresult类的一个实例,其组成很简单,为对象的基本信息,以及各基因的分数(p值或者其他统计参数
我这里随便挑一个富集结果来看看
resultFis <- runTest(GOdata, algorithm = "classic", statistic = "fisher")
-- Classic Algorithm --
the algorithm is scoring 590 nontrivial nodes
parameters:
test statistic: fisher
resultWeight <- runTest(GOdata, algorithm = "weight", statistic = "fisher")
-- Weight Algorithm --
The algorithm is scoring 590 nontrivial nodes
parameters:
test statistic: fisher : ratio
然后我们对这两种富集方式来画图
pvalFis=score(resultFis) 得到矫正的P值
pvalWeight <- score(resultWeight , whichGO = names(pvalFis))
返回resultFis和resultWeight共有的基因在resultWeight中的分数。有了这两组分数,可以做一些比较,比如关联分析:
cor(pvalFis, pvalWeight)
[1] 0.370151
library(lattice)
xyplot(pvalWeight ~ pvalFis) 画了一个散点图