昨天上午在群里看到有小伙伴问了一个很有意思的问题,就是cox生存分析结果也可以火山图可视化,而且他提问的方式值得鼓励,所以我回答了他,并且把代码分享出去了。
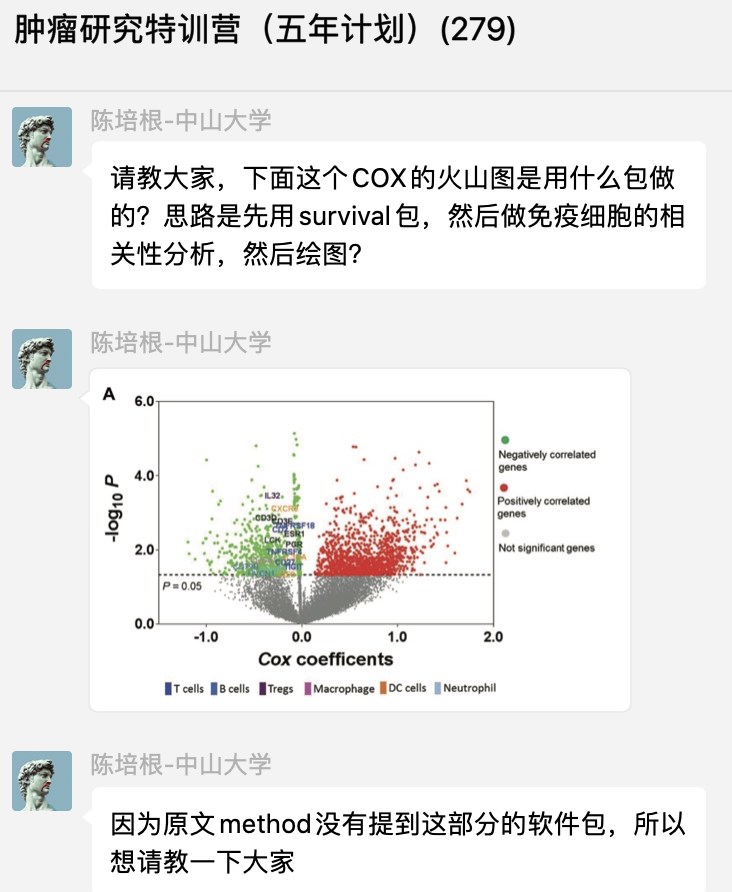
更重要的是,我觉得这个互动问答过程,值得让更多人学习,所以特意耗费半个小时整理了一个测试数据和代码,打包好了,大家只需要下载我的压缩包解压在自己电脑,就可以重复出来一个表达矩阵的全部基因的批量根据临床资料进行生存分析,cox的,而且拿到p值和HR值,并且绘制类似的火山图。
首先明白火山图的本质
其实就是一个散点图,所有基因画上去即可,每个基因是一个点,对X、Y轴有要求而已。
- X轴必须是0附近的值,比如上下调基因的变化倍数,可以log转换,这样看起来对称
- Y轴必须是统计学相关P值,进行 -log10的转换,这样与你的X轴数值离散开来就有火山喷发的感觉
差异分析当然是可以拿到这两列信息,所有基因都有logFC和 -log10P值,散点图就是我们看到的火山图啊!不过小伙伴问的是cox回归分析的结果的火山图可视化,所以我们还需要认识cox结果
然后认识cox结果
我在TCGA课程讲解过对表达矩阵的全部基因,批量做cox分析,核心代码是:
# 这里是核心代码
# 会根据每个基因的表达量中位值进行分组,然后对高低表达某基因的两组病人进行cox分析
cox_results <-apply(exprSet , 1 , function(gene){
# gene= as.numeric(exprSet[1,])
group=ifelse(gene>median(gene),'high','low')
survival_dat <- data.frame(group=group,stage=phe$stage,
stringsAsFactors = F)
m=coxph(mySurv ~ stage+ group, data = survival_dat)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['grouplow',])
})
cox_results=t(cox_results)
head(cox_results)
# 把结果保存一下,后续绘制火山图
save(cox_results,file = 'cox_results.Rdata')
拿到如下分析表格:
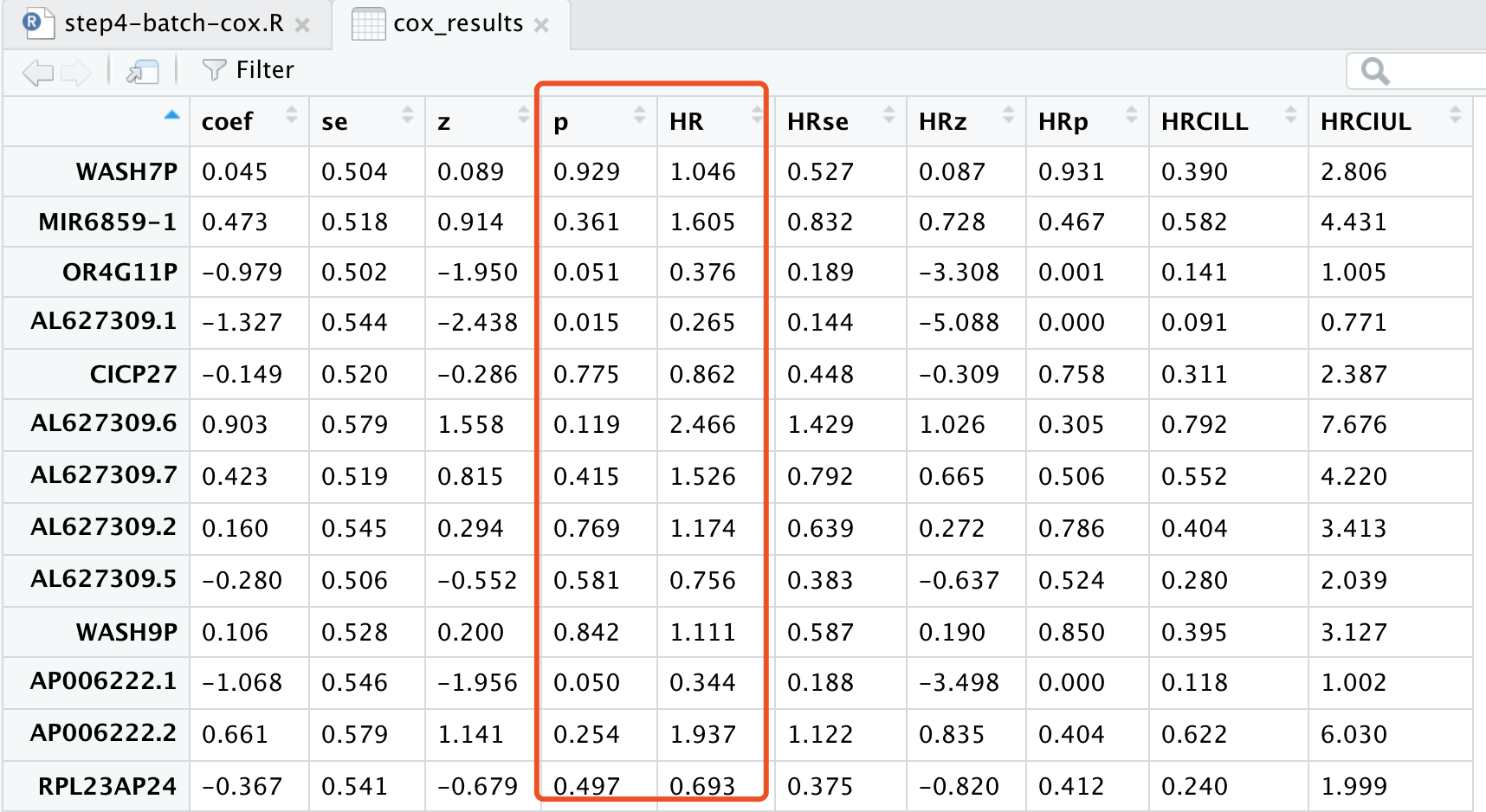
可以很清晰的看到,的确是有满足要求的两列哦。
早在三年前我就整理并且制作了TCGA肿瘤数据库知识图谱视频教程,一年半前免费公布在生信技能树的B站,现在勉勉强强也快有两万的观看量。
课程配套代码发在GitHub的TCGA视频课程上面, 如下:
step00-install-packages.R
step01-getData-from-GDC.R
step01-getData-from-RTCGA.R
step01-getData-from-Xena.R
step01-getData-from-firehose.R
step02-DEG-3-packages.R
step03-batch-logRank.R
step04-batch-coxp.R
step05-lasso.R
step06-coxph-forest.R
step07-risk-score-distribution.R
step08-Random-foreast.R
step09-miRNA-downstream.R
step10-maftools.R
step11-boxplot.R
step12-correlation.R
step13-split-cohort.R
step14-timeROC.R
step15-choose_lncRNA.R
step16-clinical-tables.R
step17-mutation-signatures.R
step17-others.R
step18-SVM.R
基本上大家看到的TCGA数据库挖掘文章里面的主流分析要点都被我的代码覆盖了,你如果有R语言基础,实际上无需看视频,直接解析我的代码就ok了。
最后对那两列数据画散点图
我们的重点并不是绘图,所以我就套用我五年前分享的GEO数据挖掘的差异基因的火山图代码:
load(file = 'cox_results.Rdata')
if(F){
need_DEG=as.data.frame(cox_results[,c(4,5)])
colnames(need_DEG)=c('pvalue','log2FoldChange')
head(need_DEG)
need_DEG$log2FoldChange=log( need_DEG$log2FoldChange)
boxplot(need_DEG$log2FoldChange)
fivenum(need_DEG$log2FoldChange)
need_DEG$log2FoldChange[abs(need_DEG$log2FoldChange)>3]=3
logFC_cutoff <- with(need_DEG,mean(abs( log2FoldChange)) + 2*sd(abs( log2FoldChange)) )
# logFC_cutoff=1
logFC_cutoff
need_DEG$change = as.factor(ifelse(need_DEG$pvalue < 0.05 & abs(need_DEG$log2FoldChange) > logFC_cutoff,
ifelse(need_DEG$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT')
)
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(need_DEG[need_DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(need_DEG[need_DEG$change =='DOWN',])
)
library(ggplot2)
g = ggplot(data=need_DEG,
aes(x=log2FoldChange, y=-log10(pvalue),
color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)
}
看起来还行,这个图:
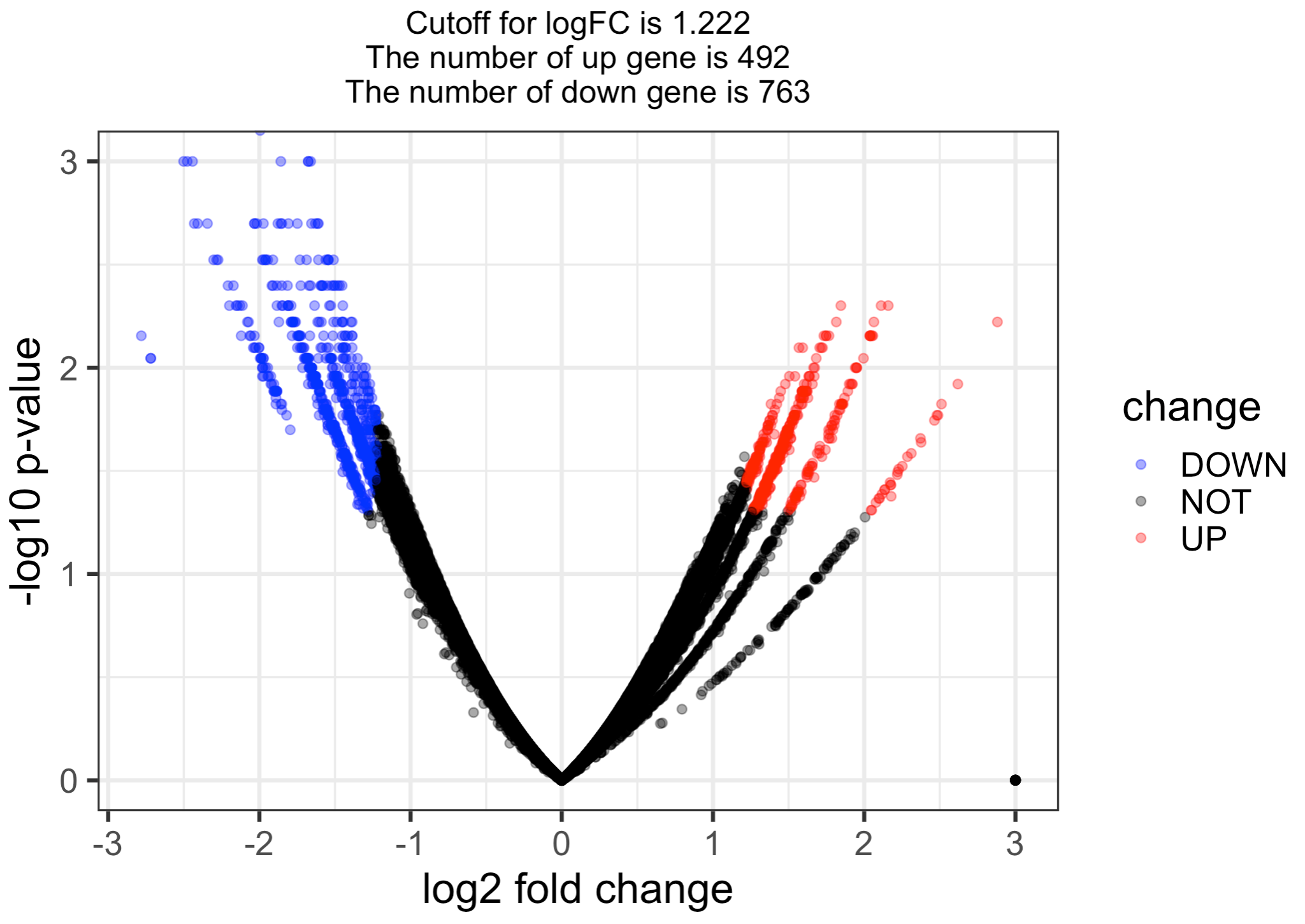
值得注意的是,这里面的logFC其实是我们的cox的HR值log哦,而且我给了一个绝对值小于3的处理。
反正我觉得这个cox结果表格的火山图可视化,勉强还行吧,至于在这个火山图上面标记基因上面的,就取决于大家自己的生物学背景咯。
付费内容分割线
为有效杜绝黑粉跟我扯皮,设置一个付费分割线,这样它们就没办法复制粘贴我的代码,也不可能给我留言骂街了!
世界顿时清净很多!
付费拿全套代码哈
代码如下,大家下载测试后,可以留言反馈,我会 根据大家的留言继续更新这个代码哈
付费方式见微信公众号推文:https://mp.weixin.qq.com/s/7RRecg5R30-jEWLK7TmT3A
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路