前面我在生信技能树分享过 批量cox生存分析结果也可以火山图可视化 介绍了火山图的基础认识,同时也给了大家代码可以批量做cox分析,并且绘制出来火山图。
最近看到一个文献,是数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101668
GSM2711785 RKO-WT-rep1
GSM2711786 RKO-WT-rep2
GSM2711787 RKO-PRDM1-KO2-rep1
GSM2711788 RKO-PRDM1-KO2-rep2
GSM2711789 RKO-PRDM1-KO5-rep1
GSM2711790 RKO-PRDM1-KO5-rep2
GSM2711791 RKO-GFP-OE-rep1
GSM2711792 RKO-GFP-OE-rep2
GSM2711793 RKO-PRDM1α-OE-rep1
GSM2711794 RKO-PRDM1α-OE-rep2
GSM2711795 RKO-PRDM1β-OE-rep1
GSM2711796 RKO-PRDM1β-OE-rep2
可以看到样本不多,但是分组不少,所以作者分析这个转录组数据的时候,可视化很丰富:
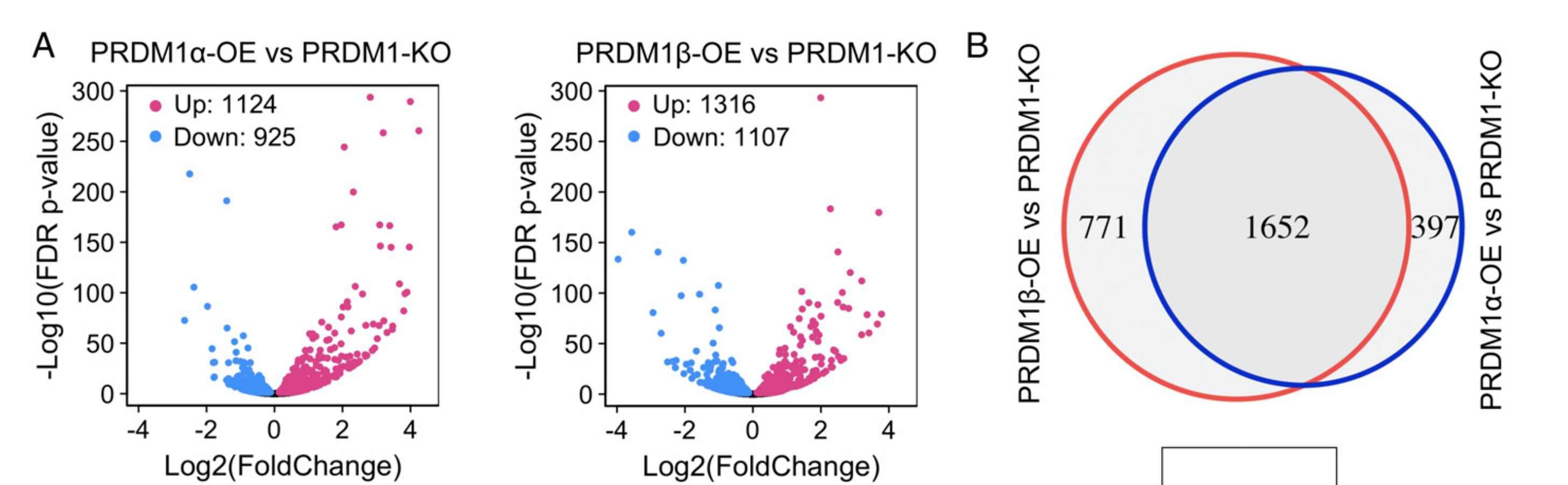
这样的火山图,韦恩图,相信大家看过我的GEO数据挖掘系列教程,肯定没有问题的啦。
我已经多次讲解了,走标准分析流程,火山图,热图,GO/KEGG数据库注释等等。这些流程的视频教程都在B站和GitHub了,目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
仅仅是最后得到的差异分子,并不是以前的mRNA后面的基因名,而是miRNA,lncRNA,甚至circRNA的ID,看起来很陌生罢了。感兴趣可以细读表达芯片的公共数据库挖掘系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
这样简单的分析,当然只能是结合生物学背景尽量去解释它。但是有一个图,值得我分享一下,就是
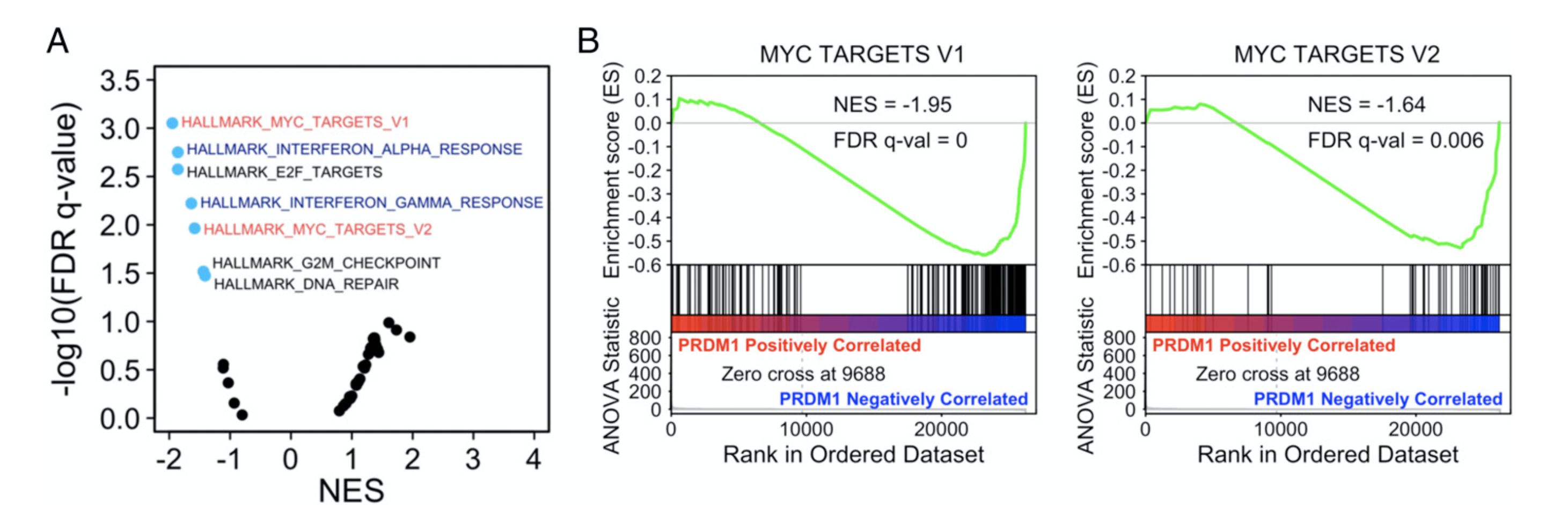
作者把表达矩阵的差异分析结果(PRDM1-KO and PRDM1-OE RKO cells.),进行gsea分析,针对MSigDB Hallmark 的50个基因集。
Volcano plot of GSEA of the MSigDB Hallmark database. The FDR versus the normalized enrichment score (NES) for each evaluated gene set is shown. Blue dots are significantly enriched gene sets (FDR adjusted P value <0.05).
当然了,针对MSigDB Hallmark 的50个基因集的gsea代码我其实也给了大家,跟我们一直讲解的针对kegg的gsea不一样哦,下面是针对kegg的:
geneList=gene$logfc
names(geneList)=gene$ENTREZID
geneList=sort(geneList,decreasing = T)
head(geneList)
library(clusterProfiler)
kk_gse <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 10,
pvalueCutoff = 0.9,
verbose = FALSE)
tmp=kk_gse@result
kk=DOSE::setReadable(kk_gse, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
然后是针对MSigDB Hallmark 的50个基因集的gsea代码,需要自己下载gmt文件哦。
#选择gmt文件(MigDB中的全部基因集)
d='~/biosoft/MSigDB/symbols/'
gmts <- list.files(d,pattern = 'all')
gmts
#GSEA分析
library(GSEABase) # BiocManager::install('GSEABase')
## 下面使用lapply循环读取每个gmt文件,并且进行GSEA分析
## 如果存在之前分析后保存的结果文件,就不需要重复进行GSEA分析。
f='gsea_results.Rdata'
if(!file.exists(f)){
gsea_results <- lapply(gmts, function(gmtfile){
# gmtfile=gmts[2]
geneset <- read.gmt(file.path(d,gmtfile))
print(paste0('Now process the ',gmtfile))
egmt <- GSEA(geneList, TERM2GENE=geneset, verbose=FALSE)
head(egmt)
# gseaplot(egmt, geneSetID = rownames(egmt[1,]))
return(egmt)
})
# 上面的代码耗时,所以保存结果到本地文件
save(gsea_results,file = f)
}
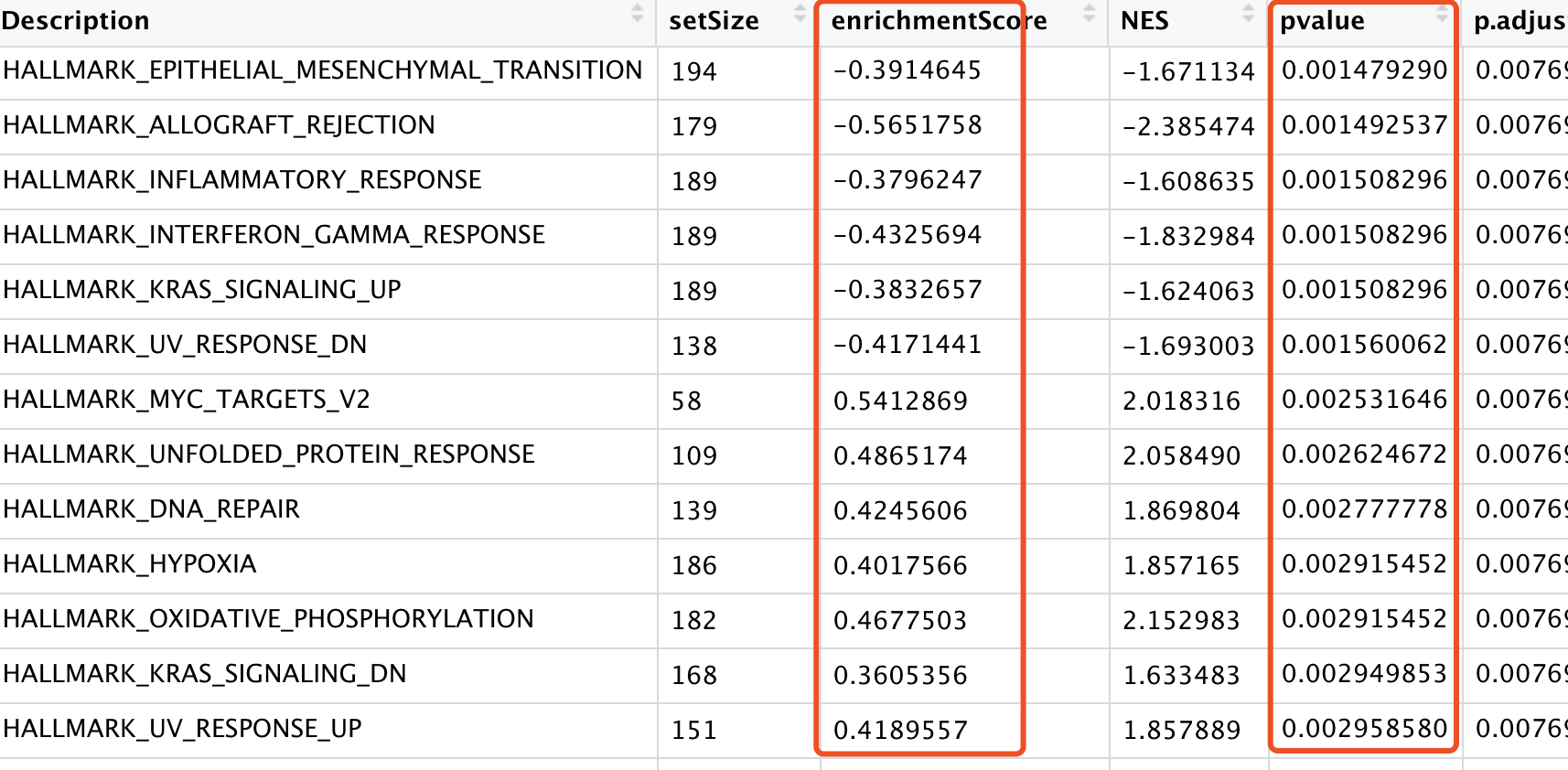
同样的,可以拿到能够被火山图展现的数据,如下:
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五),第二期 ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路