这是很久以前的一篇文章,我先贴出来给大家看看,然后讲一个实例
一:RepeatMasker的一些参数运行结果比较
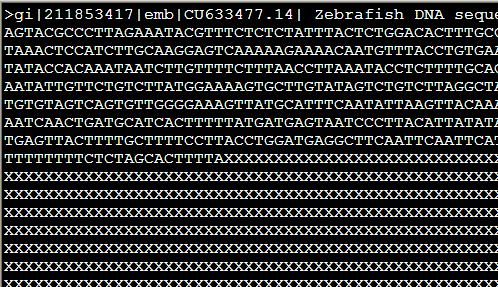
从ncbi随便下载的zebrafish的一条sequence.fasta
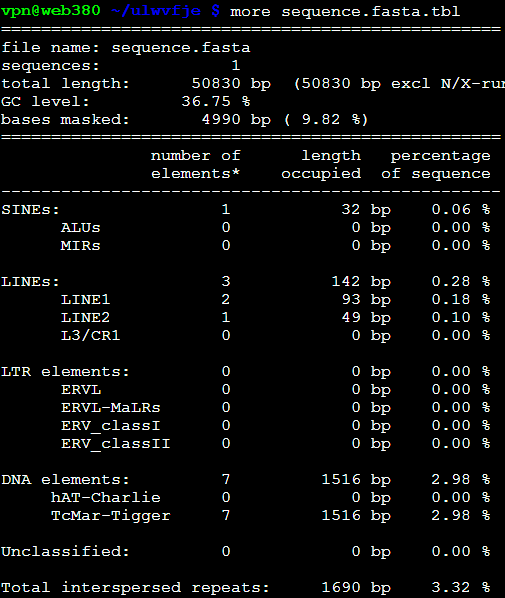
不加上任何参数跑出来结果是 RepeatMasker sequence.fasta
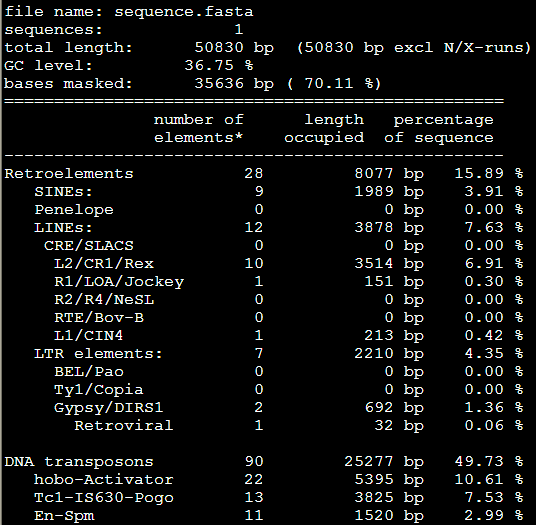
加上物种的参数之后跑出来是: RepeatMasker -species Danio sequence.fasta
效果里面出来了,之前得到的重复序列不到10%,这次可以达到70%以上,所以必须得选好对应的物种,这样才不会错过那么多要找的重复序列
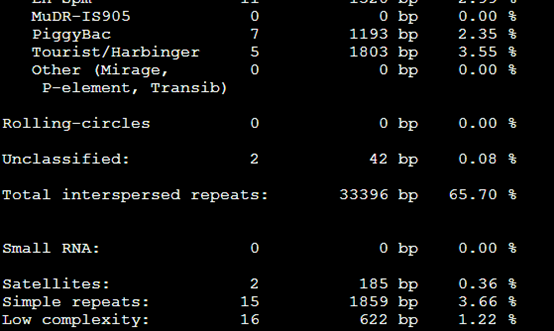
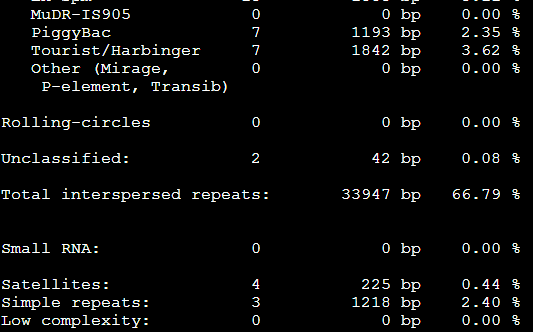
再加上-low这个参数是 RepeatMasker -species Danio -low sequence.fasta
感觉没有改变多少,就少了几个
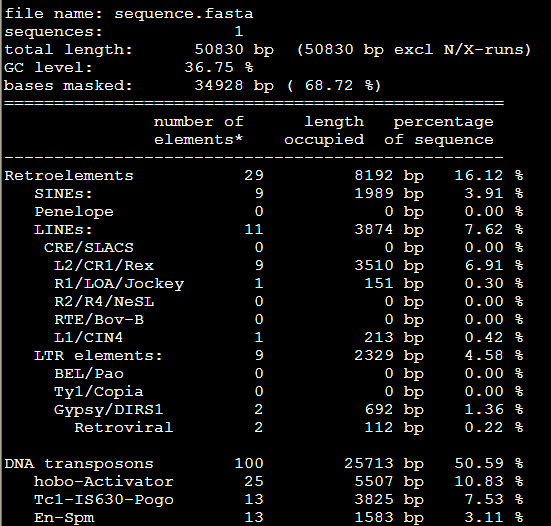
比较-div参数:RepeatMasker -species Danio sequence.fasta
RepeatMasker -species Danio -div 10 sequence.fasta
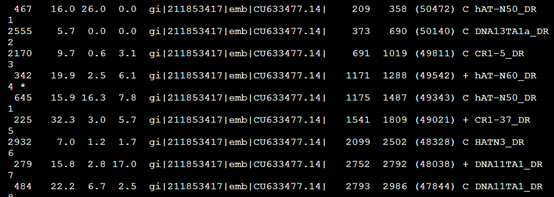
而加上-div 10之后
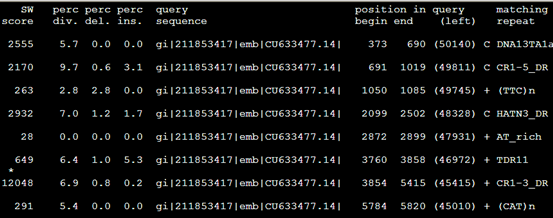
第二列小于10%的全部被剔除掉了
输出参数,本来应该是用N把重复区域屏蔽掉的
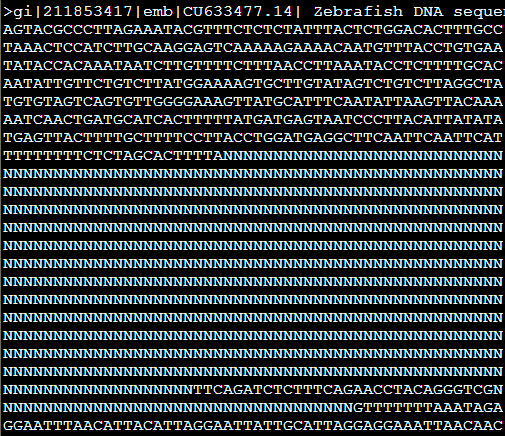
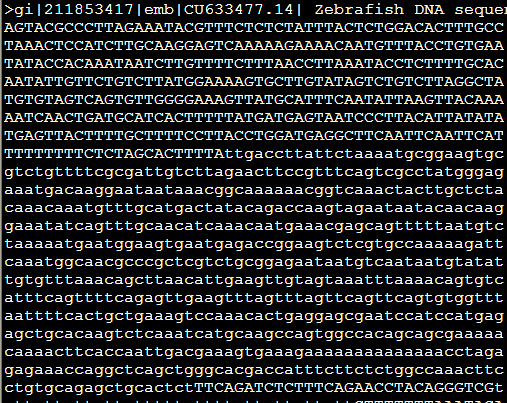
但是如果加上参数-x,原来输出是N的地方就都变成了X,感觉这个参数没啥子意义。
还有一些类似的参数,意义也不大,加上-xsmall,就是把重复区域用小写字母,不再需要N来掩盖了
如果加上-a这个参数,就多了一个文件
查看可知其内容是
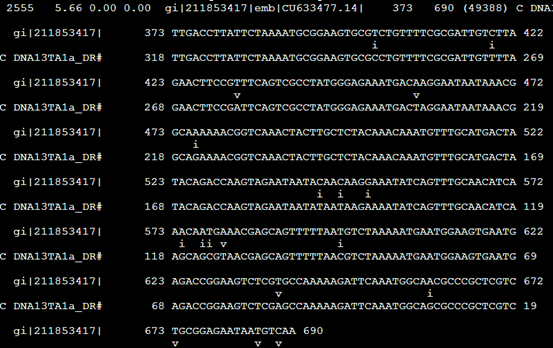
The alignments are in the cross_match/SWAT format, in which mismatches rather than matches are indicated: transitions
with an i and transversions with a v. Note it exists some differences between the alignment file and the map fi le.
The map fi le is produced by ProcessRepeats that the main task is to defragment the original map file and the alignment fi le is created from the original map fi le: the difference between them comes from the defragmented hits.
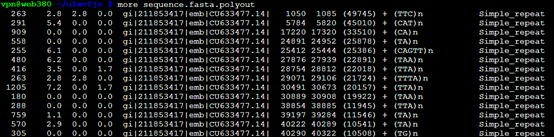
如果加上-poly,也会多出一个文件
![]()
查看,可知其单独列出了微卫星的表格
The ‘-xm’, ‘-ace,’ and ‘-gff ’ options create an additional out put file in cross match, ACeDB, and Gene Feature Finding format respectively.这几个参数都是为了生成适合其它处理的文件。
另外针对大文件的操作,可能需要-pa来设置运行速度,或者-s,-q,-qq
二:生成的文件的解释
会输出这些文件
![]()
1,。Out类文件
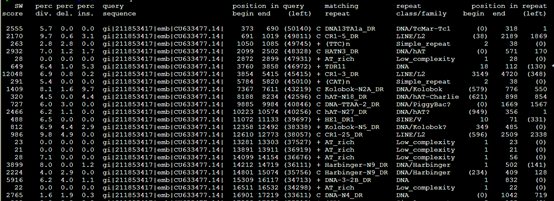
| SW score | 根据Smith-Waterman算法比对的分值 | 2555 | |
| Div% | 比上区间与共有序列相比的替代率 | 5.7 | |
| Del% | 在查询序列中碱基缺失的百分率(删除碱基) | 0.0 | |
| Ins% | 在repeat库序列中碱基缺失的百分率(插入碱基) | 0.0 | |
| Query sequence | 输入的待屏蔽重复的序列 | gi|211853417|emb|CU633477.14| | |
| Position begin | 373 | ||
| Position end | 690 | ||
| Query left | 在查询序列中超出比上区域的碱基数
+= 比上了库中重复序列的正义链,如果是互补连用“c”表示 |
(50140) | |
| Matching repeat | 比上的重复序列的名称 | C DNA13TA1a_DR | |
| Repeat family(class) | 比上的重复序列的类型 | DNA/TcMar-Tc1 | |
| Position begin | |||
| Position end | |||
| Query left | 比对区域距重复序列左端的碱基数 | ||
| 比对的顺序ID |
3.cat文件基本类似于。Out文件
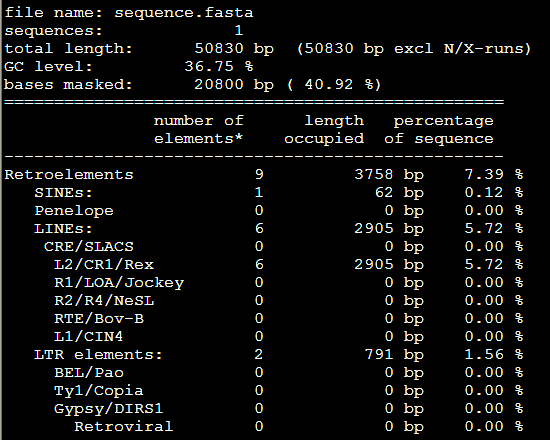
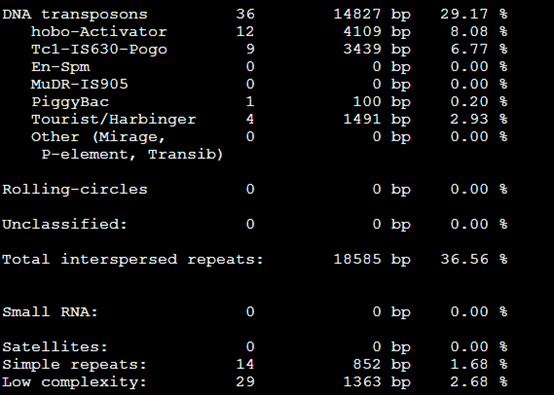
3。。Tbl类文件
4.masked文件,就是找到的重复序列被N给代替了,或者用参数改变代替形式
polyout文件。就是单独列出了微卫星表格
Align文件,其实就是把之前的。Out文件的每一行记录单独拿出来再进行表格化解释
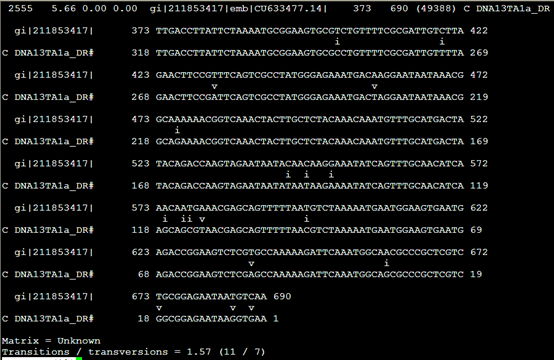
把373到690的核苷酸序列列出来,说明这个DNA13TA1a_DR 重复具体的意义
但是没看懂这个i,v是什么意思
结果比较
从ncbi随便下载的zebrafish的一条sequence.fasta
不加上任何参数跑出来结果是 RepeatMasker sequence.fasta
加上物种的参数之后跑出来是: RepeatMasker -species Danio sequence.fasta
效果里面出来了,之前得到的重复序列不到10%,这次可以达到70%以上,所以必须得选好对应的物种,这样才不会错过那么多要找的重复序列