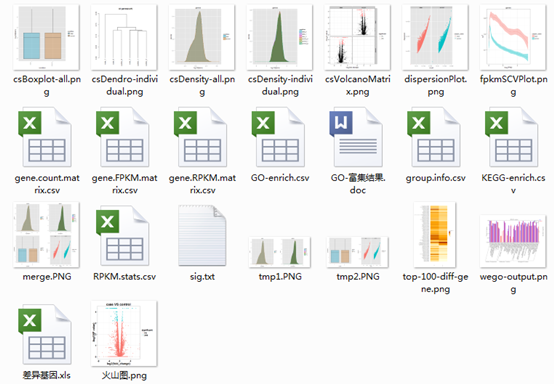
单细胞转录组数据分析CNV
都来aviv Regev自于实验室,一系列文章都利用了单细胞转录组数据分析CNV。 Continue reading
都来aviv Regev自于实验室,一系列文章都利用了单细胞转录组数据分析CNV。 Continue reading

早在去年九月,我就写个博文说 RNA-seq流程需要进化啦! http://www.bio-info-trainee.com/1022.html ,主要就是进化成hisat2+stringtie+ballgown的流程,但是我一直没有系统性的讲这个流程,因为我觉真心木有用。我只用了里面的hisat来做比对而已!但是群里的小伙伴问得特别多,我还是勉为其难的写一个教程吧,你们之间拷贝我的代码就可以安装这些软件的!然后自己找一个测试数据,我的脚本很容易用的! Continue reading
我以前写过bedtools和htseq-counts的教程,它们都可以用来对比对好的bam文件进行计数,正好群里有小伙伴问我它们的区别,我就简单做了一个比较,大家可以先看看我以前写的软件教程。写的有的挫:
言归正传,我这里没精力去探究它们的具体原理,只是看看它们数一个read是否属于某个基因的时候,区别在哪里,大家看下图: Continue reading
de novo的转录组数据,比对的时候一般用的是自己组装好的trinity.fasta序列(挑选最长蛋白的转录本序列)来做参考,用bowtie2等工具直接将原始序列比对即可。所以比对 sam/bam文件本身就包含了参考序列的每一条转录本序列ID,直接对 sam/bam文件进行counts就知道每一个基因的表达量啦!
本来我是准备自己写脚本对sam文件进行counts就好,但是发现了samtools自带这样的工具:http://www.htslib.org/doc/samtools.html
如果是针对基因组序列,那么这个功能用处不大,但是针对转录本序列,统计出来的就是我们想要的转录本表达量。 Continue reading
 http://pan.baidu.com/s/1jIvwRD8 此处找到
http://pan.baidu.com/s/1jIvwRD8 此处找到搜索可以得到非常多的流程,我这里简单分享一些,我以前搜索到的文献。
北大也有讲RNA-seq的原理
链接:http://pan.baidu.com/s/1kTmWmv9 密码:6yaz
甚至,我还有个华大的培训课程!!!这可是5天的培训教程哦,好像当初还花了五千多块钱的资料!!!
链接:http://pan.baidu.com/s/1nt5OV5B 密码:gyul
优酷也有视频,可以自己搜索看看

然后还有几个pipeline,就是生信的分析流程,即使你啥都不会,按照pipeline来也不是问题啦
export PATH=/share/software/bin:$PATH
bowtie2-build ./data/GRCh37_chr21.fa chr21
tophat -p 1 -G ./data/genes.gtf -o P460.thout chr21 ./data/P460_R1.fq ./data/P460_R2.fq
tophat -p 1 -G ./data/genes.gtf -o C460.thout chr21 ./data/C460_R1.fq ./data/C460_R2.fq
cufflinks -p 1 -o P460.clout P460.thout/accepted_hits.bam
cufflinks -p 1 -o C460.clout C460.thout/accepted_hits.bam
samtools view -h P460.thout/accepted_hits.bam > P460.thout/accepted_hits.sam
samtools view -h C460.thout/accepted_hits.bam > C460.thout/accepted_hits.sam
echo ./P460.clout/transcripts.gtf > assemblies.txt
echo ./C460.clout/transcripts.gtf >> assemblies.txt
cuffmerge -p 1 -g ./data/genes.gtf -s ./data/GRCh37_chr21.fa assemblies.txt
cuffdiff -p 1 -u merged_asm/merged.gtf -b ./data/GRCh37_chr21.fa -L P460,C460 -o P460-C460.diffout P460.thout/accepted_hits.bam C460.thout/accepted_hits.bam
samtools index P460.thout/accepted_hits.bam
samtools index C460.thout/accepted_hits.bam
和另外一个
#!/bin/bash
# Approx 75-80m to complete as a script
cd ~/RNA-seq
ls -l data
tophat --help
head -n 20 data/2cells_1.fastq
time tophat --solexa-quals \
-g 2 \
--library-type fr-unstranded \
-j annotation/Danio_rerio.Zv9.66.spliceSites\
-o tophat/ZV9_2cells \
genome/ZV9 \
data/2cells_1.fastq data/2cells_2.fastq # 17m30s
time tophat --solexa-quals \
-g 2 \
--library-type fr-unstranded \
-j annotation/Danio_rerio.Zv9.66.spliceSites\
-o tophat/ZV9_6h \
genome/ZV9 \
data/6h_1.fastq data/6h_2.fastq # 17m30s
samtools index tophat/ZV9_2cells/accepted_hits.bam
samtools index tophat/ZV9_6h/accepted_hits.bam
cufflinks --help
time cufflinks -o cufflinks/ZV9_2cells_gff \
-G annotation/Danio_rerio.Zv9.66.gtf \
-b genome/Danio_rerio.Zv9.66.dna.fa \
-u \
--library-type fr-unstranded \
tophat/ZV9_2cells/accepted_hits.bam # 2m
time cufflinks -o cufflinks/ZV9_6h_gff \
-G annotation/Danio_rerio.Zv9.66.gtf \
-b genome/Danio_rerio.Zv9.66.dna.fa \
-u \
--library-type fr-unstranded \
tophat/ZV9_6h/accepted_hits.bam # 2m
# guided assembly
time cufflinks -o cufflinks/ZV9_2cells \
-g annotation/Danio_rerio.Zv9.66.gtf \
-b genome/Danio_rerio.Zv9.66.dna.fa \
-u \
--library-type fr-unstranded \
tophat/ZV9_2cells/accepted_hits.bam # 16m
time cufflinks -o cufflinks/ZV9_6h \
-g annotation/Danio_rerio.Zv9.66.gtf \
-b genome/Danio_rerio.Zv9.66.dna.fa \
-u \
--library-type fr-unstranded \
tophat/ZV9_6h/accepted_hits.bam # 13m
time cuffdiff -o cuffdiff/ \
-L ZV9_2cells,ZV9_6h \
-T \
-b genome/Danio_rerio.Zv9.66.dna.fa \
-u \
--library-type fr-unstranded \
annotation/Danio_rerio.Zv9.66.gtf \
tophat/ZV9_2cells/accepted_hits.bam \
tophat/ZV9_6h/accepted_hits.bam # 7m
head -n 20 cuffdiff/gene_exp.diff
sort -t$'\t' -g -k 13 cuffdiff/gene_exp.diff \
> cuffdiff/gene_exp_qval.sorted.diff
head -n 20 cuffdiff/gene_exp_qval.sorted.diff
网站成立也快一个月了,总算是完全搞定了生信领域的一个方向,当然,只是在菜鸟层面上的搞定,还有很多深层次的应用及挖掘,仅仅是我所讲解的这些软件也有多如羊毛的参数可以变幻,复杂的很。其实我最擅长的并不是转录组,但是因为一些特殊的原因,我恰好做了三个转录组项目,所以手头上关于它的资料比较多,就分享给大家啦!稍后我会列一个网站更新计划,就好谈到我所擅长的基因组及免疫组库。我这里简单对转录组做一个总结:
首先当然是我的转录组分类网站啦
http://www.bio-info-trainee.com/?cat=18
同样的我用脚本总结一下给大家
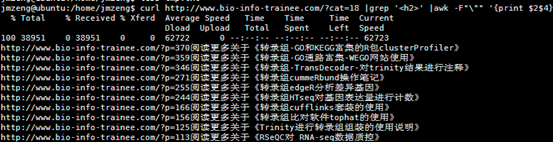
http://www.bio-info-trainee.com/?p=370阅读更多关于《转录组-GO和KEGG富集的R包clusterProfiler》
http://www.bio-info-trainee.com/?p=359阅读更多关于《转录组-GO通路富集-WEGO网站使用》
http://www.bio-info-trainee.com/?p=346阅读更多关于《转录组-TransDecoder-对trinity结果进行注释》
http://www.bio-info-trainee.com/?p=271阅读更多关于《转录组cummeRbund操作笔记》
http://www.bio-info-trainee.com/?p=255阅读更多关于《转录组edgeR分析差异基因》
http://www.bio-info-trainee.com/?p=244阅读更多关于《转录组HTseq对基因表达量进行计数》
http://www.bio-info-trainee.com/?p=166阅读更多关于《转录组cufflinks套装的使用》
http://www.bio-info-trainee.com/?p=156阅读更多关于《转录组比对软件tophat的使用》
http://www.bio-info-trainee.com/?p=125阅读更多关于《Trinity进行转录组组装的使用说明》
http://www.bio-info-trainee.com/?p=113阅读更多关于《RSeQC对 RNA-seq数据质控》
同时我也讲了如何下载数据
http://www.bio-info-trainee.com/?p=32
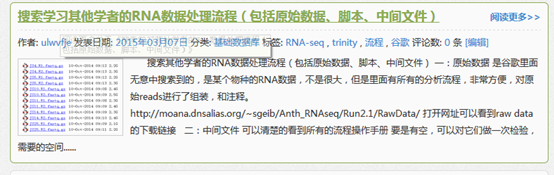
原始SRA数据首先用SRAtoolkit数据解压,然后进行过滤,评估质量,然后trinity组装,然后对组装好的进行注释,然后走另一条路进行差异基因,差异基因有tophat+cufflinks+cummeRbund,也有HTseq 和edgeR等等,然后是GO和KEGG通路注释,等等。
在我的群里面共享了所有的代码及帖子内容,欢迎加群201161227,生信菜鸟团!
http://www.bio-info-trainee.com/?p=1
线下交流-生物信息学
同时欢迎下载使用我的手机安卓APP
http://www.cutt.com/app/down/840375
转录组cummeRbund操作笔记
这是跟tophat和cufflinks套装紧密搭配使用的一个R包,能出大部分文章要求的标准化图片。
一:安装并加装该R包
安装就用source("http://bioconductor.org/biocLite.R") ;biocLite("cummeRbund")即可,如果安装失败,就需要自己下载源码包,然后安装R模块。
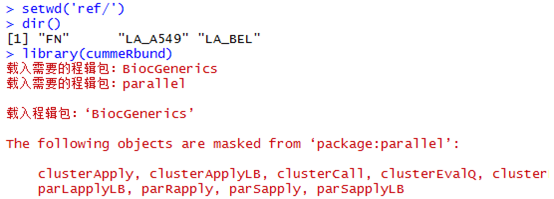
然后把cuffdiff输出的文件目录拷贝到R的工作目录,或者自己设置工作目录
二:读取FN目录下面的所有文件。
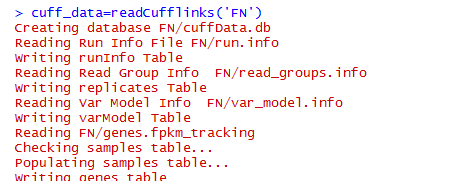
可以看到把cuffdiff下面的文件夹所有的文件都读取到了,里面有如下文件,包括genes,isoforms,cds,tss这四种差异情况都读取了。
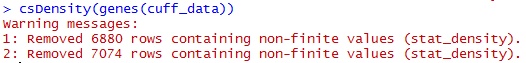
三:表达水平分布图
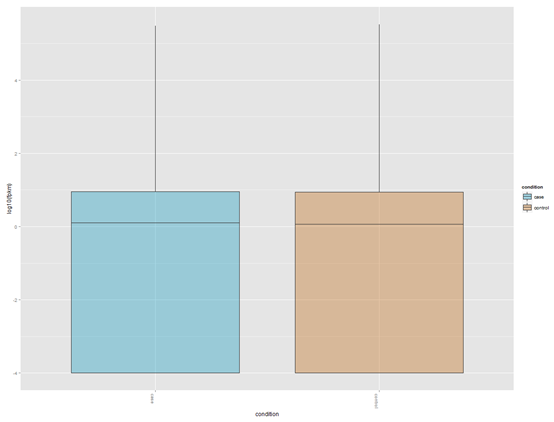
四、表达水平箱线图
csBoxplot(genes(cuff_data))
五、画基因表达差异热图
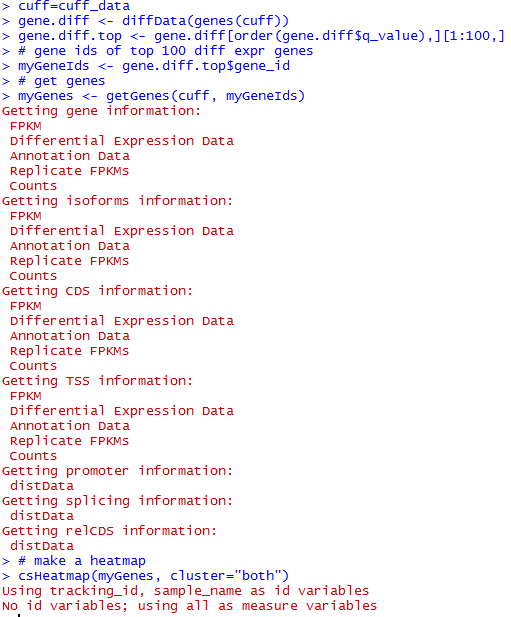
画出热图如下
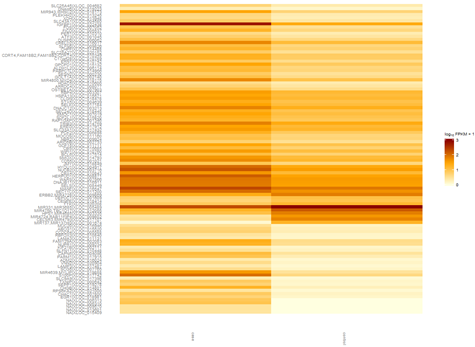
六、得到差异的genes,isoforms,TSS,CDS等等
diffData <- diffData(myGenes )

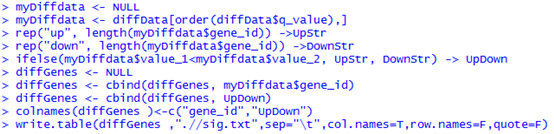
只有一百个有表达差异的基因
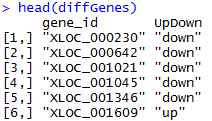
最后贴出一个综合性的代码,算了,太浪费空间了,把整个空间搞得不好看,就不贴了。
这个代码可以自动运行出图;