基本上每年我们都会在生信技能树等公众号写多个教程分享WGCNA的实战细节,比如:
- 一文看懂WGCNA 分析(2019更新版) (点击阅读原文即可拿到测序数据)
- 通过WGCNA作者的测试数据来学习
- 重复一篇WGCNA分析的文章(代码版)
- 重复一篇WGCNA分析的文章(解读版)(逆向收费读文献2019-19)
- 关键问题答疑:WGCNA的输入矩阵到底是什么格式
- WGCNA-流程及原理细节直播互动授课
代码也是完全公开的,大家很容易复制粘贴到自己的表达量矩阵群,其实算起来WGCNA本身就一个函数而已,就是划分基因模块,其它都是附加分析。总体来说就是4个步骤:
- 合适的矩阵(一般来说,是多个基因或者其它特征指标在多个样品的矩阵)
- 运行WGCNA,对基因等特征划分模块
- 计算模块和样品其它属性的相关性
- 对模块进行go或者kegg等数据库注释搞清楚模块的功能
下面我们就针对前面的 院士课题组的WGCNA数据挖掘文章能复现吗 进行4步骤复现:
合适的矩阵
前面的 院士课题组的WGCNA数据挖掘文章能复现吗 教程里面,我们拿到了转录组差异分析后的上下调基因列表,然后目前的转录组测序表达量矩阵里面的基因表达量是整数格式的counts值,并不适合做后续的运行WGCNA,对基因等特征划分模块。需要一个简单的转换,代码如下所示:
rm(list = ls())
load( file = '../step1-deg/DEG_deseq2.Rdata' )
colnames(DEG_deseq2)
nrDEG=DEG_deseq2[,c("log2FoldChange", "padj")]
head(nrDEG)
colnames(nrDEG)=c('logFC','P.Value')
# 凡是阈值,都是可以自定义
logFC_t <- 2.5
pvalue_t <- 0.001
gene_up= rownames( nrDEG[with(nrDEG,
logFC > logFC_t & P.Value < pvalue_t),])
gene_down=rownames( nrDEG[with(nrDEG,
logFC < -logFC_t & P.Value < pvalue_t),])
length(gene_up);length(gene_down)
head(gene_up);head(gene_down)
load(file = '../step1-deg/symbol_matrix.Rdata')
symbol_matrix[1:4,1:4] ## 基因名字的样品,矩阵
dat = log2(edgeR::cpm(symbol_matrix)+1)
# 这个归一化方法不太推荐
library(limma)
library(edgeR)
library(DESeq2)
dge <- DGEList(counts=symbol_matrix)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(symbol_matrix)
design
v <- voom(dge,design,plot=TRUE, normalize="quantile")
dat = v$E
dat[1:4,1:4]
boxplot(dat,las=2)
table(group_list)
kp = group_list=='case'
# 矩阵,样品过滤,基因过滤
dat = dat[c(gene_up,gene_down),kp]
dim(dat)
仅仅是因为前面的 院士课题组的WGCNA数据挖掘文章能复现吗 ,它是先差异分析,然后做wgcna,并不代表我们一定要这样做,如何过滤基因都是可以有自己的考虑,言之有理即可,也可以是简简单单基因sd或者mad的排序后取top 5000或者其它数量级的基因列表做后续的wgcna
然后运行WGCNA,对基因等特征划分模块
运行WGCNA之前需要确定一下软阈值,当然了,这个步骤也是可以自由修改的,
#1.确定软阈值----
library(WGCNA)
dat0 <- t(dat)
powers = c(c(1:10), seq(from = 12, to=20, by=2))
# Call the network topology analysis function
sft = pickSoftThreshold(dat0,
powerVector = powers,
verbose = 5)
po <- sft$powerEstimate;po
# Plot the results:
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1],
-sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",
ylab="Scale Free Topology Model Fit,signed R^2",
type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1],
-sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1],
sft$fitIndices[,5],
xlab="Soft Threshold (power)",
ylab="Mean Connectivity",
type="n",main = paste("Mean connectivity"))
text(sft$fitIndices[,1],
sft$fitIndices[,5],
labels=powers, cex=cex1,col="red")
#2.一步构建网络----
# 报错:https://blog.csdn.net/liyunfan00/article/details/91686840
cor <- WGCNA::cor
net = blockwiseModules(dat0, power = po,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = F,
verbose = 3) #一步构建网络
#改回去
cor<-stats::cor
class(net)
names(net)
table(net$colors)
#(1)保存net相关信息
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(net,MEs, moduleLabels, moduleColors, geneTree,
file = "Step2networkConstruction-auto.RData")
可以看到,模块划分的非常棒啊:
可以看到是10个模块,每个模块都有几百个基因 :
0 1 2 3 4 5 6 7 8 9
509 993 515 341 290 272 209 197 160 122
简简单单的 blockwiseModules 函数就完成了wgcna分析
计算模块和样品其它属性的相关性
模块是基因的合集,它可以有自己的值,也是跟基因一样的在每个样品都有一个计算好的值。同时呢,每个样品也有其它属性,比如病人就可以有年龄,体重等等,肿瘤病人还有tnm和stage信息,比如下面的代码,我们选择了肿瘤病人的转移与否这个二分类变量,取跟前面的模块进行关联分析
#1.Modules-traits relationships----
# Define numbers of genes and samples
dat <- t(dat)
nGenes = ncol(dat)
nSamples = nrow(dat)
# Recalculate MEs with color labels
MEs0 = moduleEigengenes(dat, moduleColors)$eigengenes
MEs0[1:6,1:6]
MEs = orderMEs(MEs0)
MEs[1:6,1:6]
phe = data.table::fread('TCGA-CHOL.GDC_phenotype.tsv.gz',data.table = F)
colnames(phe)
phe=phe[match(rownames(dat),phe$submitter_id.samples),]
# [41] "pathologic_M"
# [42] "pathologic_N"
# [43] "pathologic_T"
group_list = phe$pathologic_M
table(group_list)
group_list=ifelse(group_list=='M0','non','M')
group_list=as.factor(group_list)
# 肿瘤,tmn,stage。。。。
design=model.matrix(~0+ group_list)
colnames(design)=levels(group_list)
moduleTraitCor = cor(MEs, design, use = "p")
head(moduleTraitCor)
moduleTraitPvalue = corPvalueStudent(moduleTraitCor,
nSamples)
head(moduleTraitPvalue)
#可视化
sizeGrWindow(10,6)
# Will display correlations and their p-values
textMatrix = paste(signif(moduleTraitCor, 2),
"\n(",signif(moduleTraitPvalue, 1),
")",
sep = "");
dim(textMatrix) = dim(moduleTraitCor)
textMatrix[1:6,1:3]
par(mar = c(3, 12, 3, 1));
# Display the correlation values within a heatmap plot
design1 <- as.data.frame(design)
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(design1),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = blueWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 1,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
可以看到,有两个模块跟我们的肿瘤转移是正相关,一个模块是负相关,如下所示:
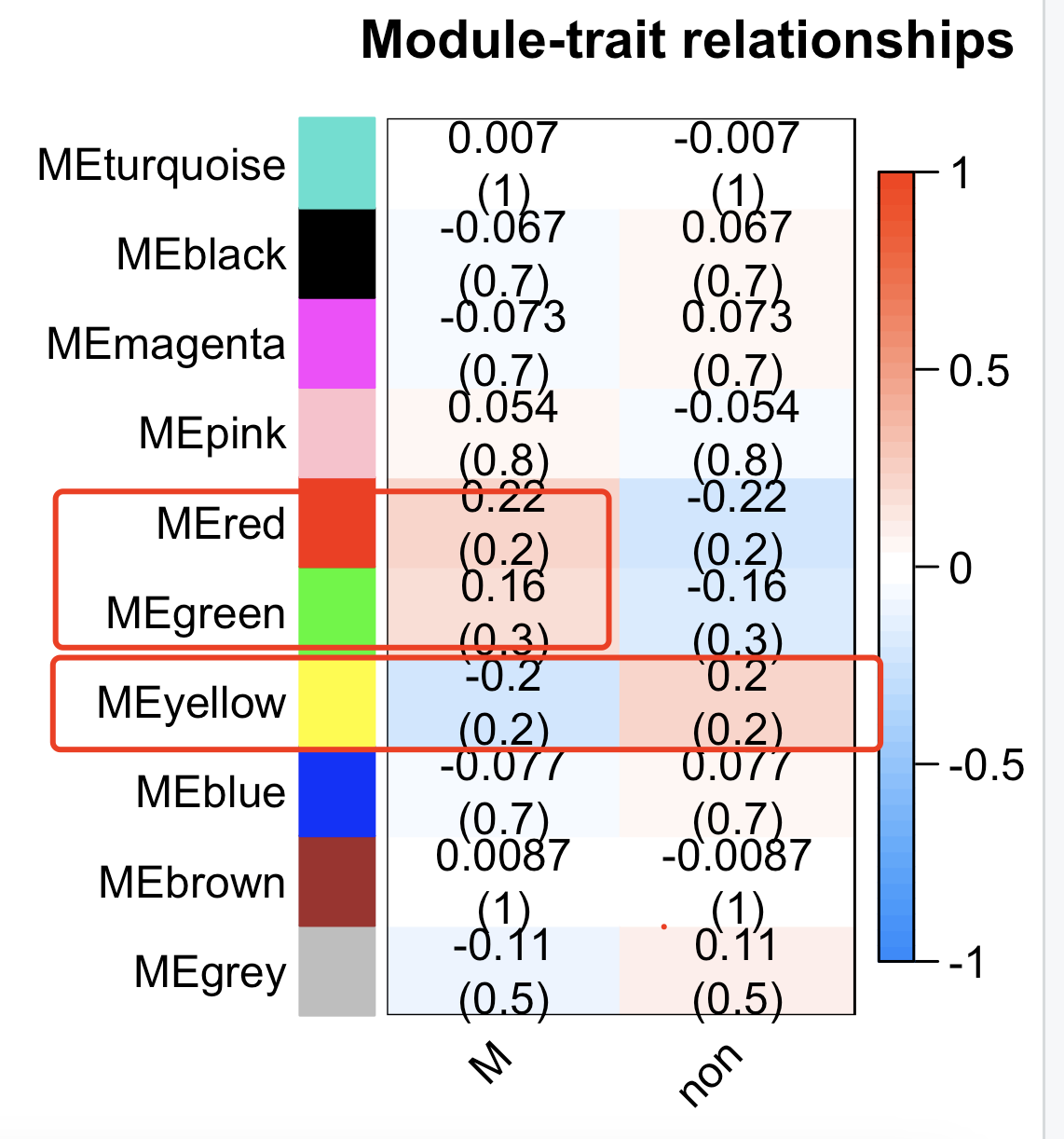
最后就是对模块进行go或者kegg等数据库注释搞清楚模块的功能
前面我们知道了有两个模块跟我们的肿瘤转移是正相关,一个模块是负相关,而且也知道了模块里面的基因是什么,但是基因那么多我们没办法描述它,通常是需要对各个模块里面的基因进行go或者kegg等数据库注释搞清楚模块的功能。代码也是很简单的:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(WGCNA)
load( file = "Step2networkConstruction-auto.RData")
table(moduleColors)
group_g=data.frame(gene=names(net$colors),
group=moduleColors)
head(group_g)
#save(group_g,file='wgcna_group_g.Rdata')
library(clusterProfiler)
# Convert gene ID into entrez genes
head(group_g)
tmp <- bitr(group_g$gene, fromType="SYMBOL",
toType="ENTREZID",
OrgDb="org.Hs.eg.db")
de_gene_clusters=merge(tmp,group_g,by.x='SYMBOL',by.y='gene')
table(de_gene_clusters$group)
head(de_gene_clusters)
gcSample <- split(de_gene_clusters$ENTREZID,
de_gene_clusters$group)
xx <- compareCluster(gcSample, fun="enrichKEGG",
organism="hsa", pvalueCutoff=0.05)
dotplot(xx)
如下所示,我们不仅仅是是知道了有两个模块跟我们的肿瘤转移是正相关,而且知道了这两个模块的功能是细胞恶性增殖以及细胞迁移,完美的对应了肿瘤的恶性转移。
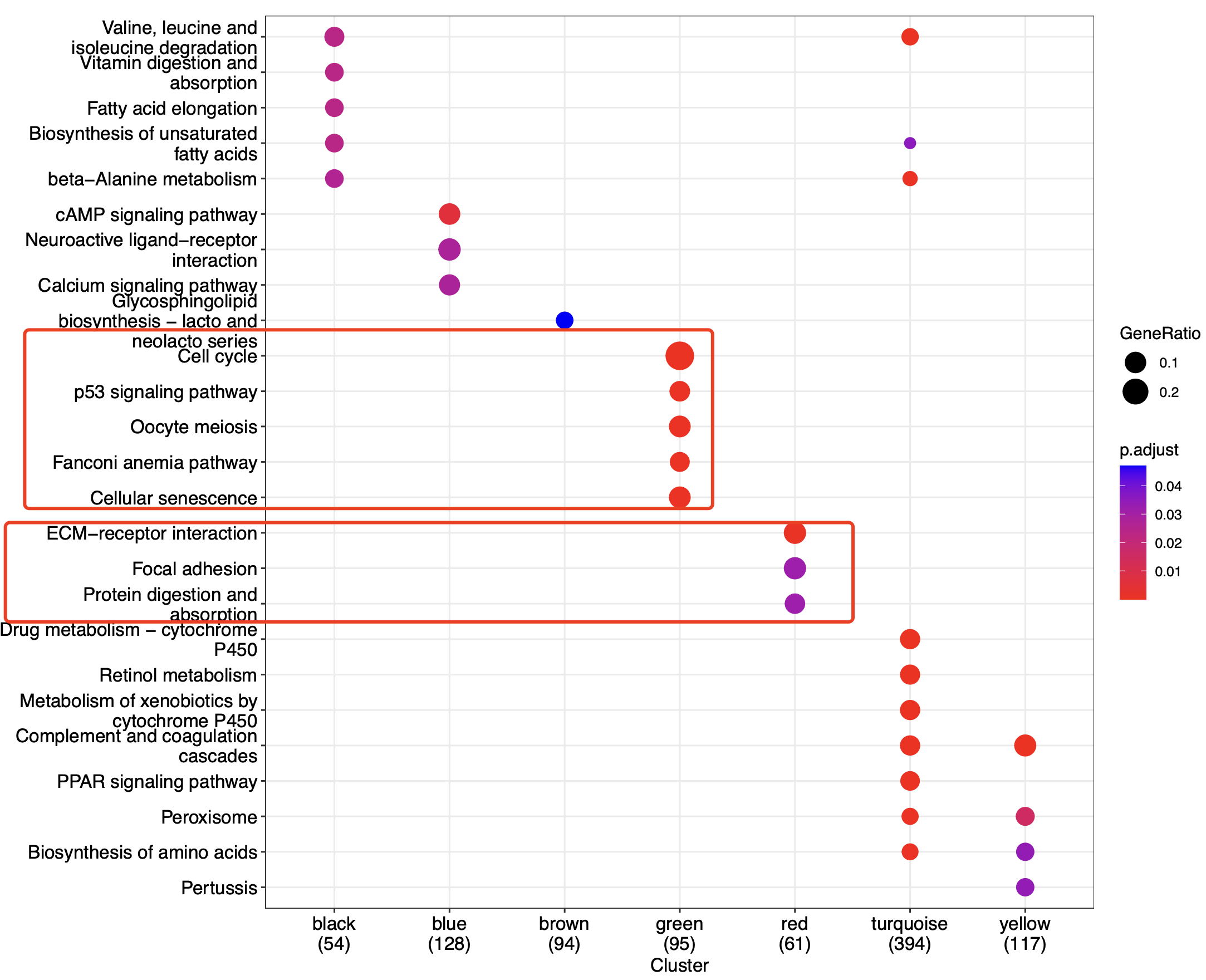
我们这里并没有去重复院士团队的WGCNA挖掘文章,而是自作主张的把它修改成为癌症转移与否关键模块,结果也挺好的。
相比大家也懂我们为什么没有去重复院士团队的WGCNA挖掘文章的细节。代码这里给大家,感兴趣的可以去读一下,蛮有意思的:https://cowtransfer.com/s/9c88947d74bf4b 点击链接查看 [ 2023-浙江大学李兰娟院士-WGCNA数据挖掘.zip ] ,或访问奶牛快传 cowtransfer.com 输入传输口令 whhffg 查看;