前面我们直播了转录组测序后的表达量矩阵的下游分析标准代码,其中为了方便大家复现,我们使用了人类的airway数据集,它表达量矩阵整理代码如下所示:
# 魔幻操作,一键清空
rm(list = ls())
options(stringsAsFactors = F)
library(data.table)
library(airway,quietly = T)
data(airway)
mat <- assay(airway)
mat[1:4,1:4]
keep_feature <- rowSums (mat > 1) > 1
ensembl_matrix <- mat[keep_feature, ]
library(AnnoProbe)
head(rownames(ensembl_matrix))
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
head(ids)
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
symbol_matrix[1:4,1:4]
group_list = as.character(airway@colData$dex);group_list
group_list=ifelse(group_list=='untrt','control','case' )
table(group_list)
group_list = factor(group_list,levels = c('control','case' ))
save(symbol_matrix,group_list,file = 'symbol_matrix.Rdata')
值得注意的是,里面的 AnnoProbe包是可以根据不同物种的ENSEMBL信息去转为SYMBOL信息,实际上它这个转换是基于我对人类和小鼠的gtf文件的解析。
关于GTF(Gene Transfer Format)文件格式
它是一种常用于存储基因组注释信息的文本文件格式。它被广泛用于描述基因的结构、外显子、内含子以及其他与基因相关的生物学特征。GTF文件格式通常用于将基因组注释信息传递给生物信息学工具和数据库,以便于基因识别、功能预测和基因组分析。
以下是GTF文件格式的基本结构和注释信息内容:
- 基本结构: GTF文件由多行文本组成,每一行代表一个注释的特征,如基因、转录本、外显子等。每一行被分成多个字段,这些字段通过制表符(Tab)或空格进行分隔。
- 字段信息: 通常,GTF文件的每一行都包含以下字段:
- 染色体编号(Chromosome): 特征所在的染色体。
- 数据源(Source): 提供注释信息的数据来源,例如数据库或软件。
- 特征类型(Feature type): 描述特征的类型,如基因、转录本、外显子等。
- 起始位置(Start): 特征在染色体上的起始位置。
- 终止位置(End): 特征在染色体上的终止位置。
- 分数(Score): 特征的质量得分,通常为浮点数。
- 链向性(Strand): 特征所在链的方向,可以是正链(+)或负链(-)。
- 相位(Frame): 对于具有相位的特征(例如外显子),指定其在基因组上的相对位置。
- 属性(Attributes): 包含附加信息的字段,通常是一个键值对列表,提供关于特征的更多详细信息。
- 属性字段: 属性字段(Attributes)是GTF文件中的一个重要部分,它包含用于描述特征的附加信息。常见的属性包括基因名、转录本ID、外显子编号等。这些属性有助于标识和关联不同的特征。
GTF文件格式的示例:
chr1 HAVANA gene 11869 14412 . + . gene_id "ENSG00000223972"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene";
chr1 HAVANA transcript 11869 14409 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana";
chr1 HAVANA exon 11869 12227 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana"; exon_number "1"; exon_id "ENSE00002234944"; exon_version "1"; tag "basic"; transcript_support_level "1";
在生物信息学中,GTF文件格式用于存储基因组注释信息,这些信息对于基因识别、基因功能预测、转录本分析和其他相关研究非常重要。
但是我上面的示范代码里面仅仅是针对人类和小鼠,因为他们的gtf文件我解析了。这个时候交流群的小伙伴求助需要解析非洲绿猴的gtf文件,下载得到的文件如下所示:
180M Aug 18 15:42 Chlorocebus_sabaeus.ChlSab1.1.110.gtf*
之前我们有教程告诉大家如何使用Linux的文本处理代码,或者R语言里面的包,进行处理:
- 使用refGenome加上dplyr玩转gtf文件
- 从一个被更新后的GTF文件得到geneID和gene类型的对应关系
但是这些教程都需要自己从零开始解析gtf文件规则,然后写个性化代码。但是现在有了新选择,就是我们的中国区chatGPT解析gtf文件:
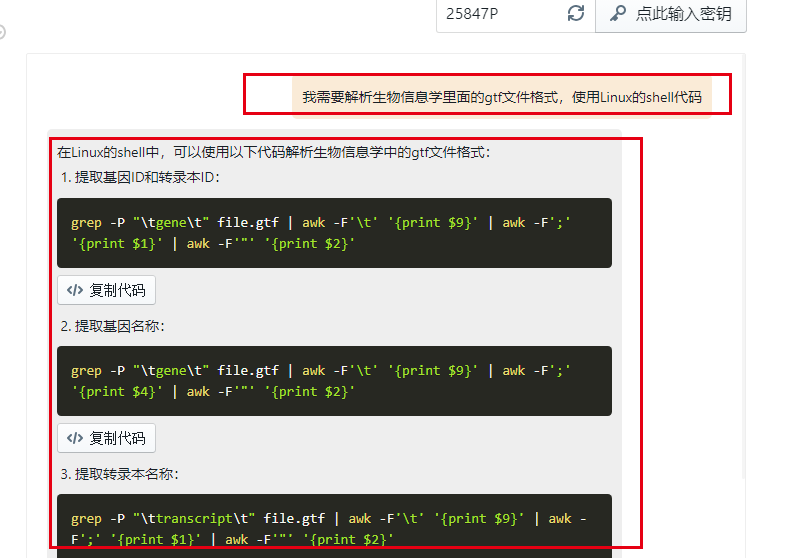
可以看到,我仅仅是提问:我需要解析生物信息学里面的gtf文件格式,使用Linux的shell代码?
它就给了我十几个小技巧,代码基本上都是可以使用的,而且Linux文本处理水平是超过我的。但是我需要的是我提取的是提取基因ID和基因名字的对应关系,所以我再次询问了它
grep -P "\tgene\t" file.gtf | awk -F'\t' '{print $9}' | awk -F';' '{print $1,$4}' | awk -F'"' '{print $2,$4}'
虽然它给我的代码是错误的,但是基本的框架是没有问题的,只需要简简单单修改print函数后面的列的编号即可,它给出来的例子是第1和4列,实际上如果是需要该物种的ENSEMBL信息去转为SYMBOL信息,得第3列:
ln Chlorocebus_sabaeus.ChlSab1.1.110.gtf file.gtf
grep -P "\tgene\t" file.gtf | awk -F'\t' '{print $9}' | awk -F';' '{print $1,$4}' | awk -F'"' '{print $2,$4}' >id2type.txt
grep -P "\tgene\t" file.gtf | awk -F'\t' '{print $9}' | awk -F';' '{print $1,$3}' | awk -F'"' '{print $2,$4}' > id2symbol.txt
也就是说,只需要简简单单Linux文本处理的基本语法,哪怕是代码学的并不好,在chatGPT的辅助下我们很容易解决问题。
在Linux环境下,AWK是一种强大的文本处理工具,用于对结构化文本文件进行数据提取、转换和报告生成等操作。它特别适用于处理以行为单位的结构化数据,如表格、日志文件等。以下是AWK语法的基本概述:
基本语法结构:
bashCopy code
awk 'pattern { action }' input_file
pattern:用于匹配要处理的行的条件,可以是正则表达式、比较表达式等。如果未提供pattern,则默认匹配所有行。action:在匹配到满足条件的行时要执行的操作,可以是对行的操作、变量赋值、打印等。
常用内置变量:
$0:表示整行内容。$1,$2, …:表示分隔后的字段,以空格或制表符为分隔符。NF:表示字段数量。NR:表示当前行号。FS:表示字段分隔符,默认为制表符。
示例操作:
# 打印文件的每一行
awk '{ print }' input.txt
# 打印文件的第二列
awk '{ print $2 }' input.txt
# 打印包含"keyword"的行
awk '/keyword/ { print }' input.txt
# 计算并打印文件的行数
awk 'END { print NR }' input.txt
示例应用:
# 提取CSV文件的第三列并计算总和
awk -F ',' '{ sum += $3 } END { print sum }' data.csv
# 打印文件中字段数量大于等于3的行
awk 'NF >= 3 { print }' input.txt
# 打印每行的第一个和最后一个字段
awk '{ print $1, $NF }' input.txt
# 格式化打印,将第一列的内容作为键,第二列的内容作为值
awk '{ data[$1] = $2 } END { for (key in data) print key, data[key] }' input.txt
这些示例只是AWK功能的一小部分,AWK还支持更多高级的操作,如条件控制、循环、函数等。AWK在文本处理中非常有用,可以帮助您高效地从结构化文本文件中提取有用的信息、执行计算和生成报告。
虽然我们在chatGPT的辅助下,搞该物种的ENSEMBL信息去转为SYMBOL信息是大体上没有问题了,但是因为这样的物种本来就是不同数据库信息会有冲突,因为不同数据库来源于不同机构,如下所示:
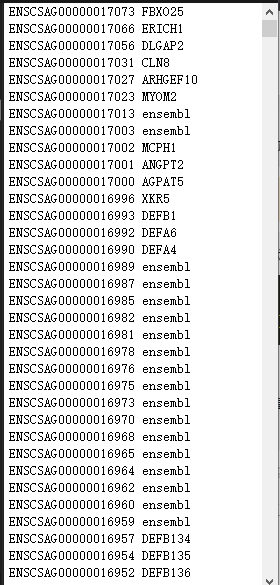
如果去一个个检查那先失败的id转换,可以看到其实是因为它本来就是没有ensembl对应的symbol,所以失败才是理所当然的。
8 ensembl gene 7020215 7022291 . + .
gene_id "ENSCSAG00000016964"; gene_version "1"; gene_source "ensembl"; gene_biotype "protein_coding";
在生物信息学中,将ENSEMBL ID(例如基因、转录本、蛋白质等)转换为SYMBOL(通常是基因的官方符号或别名)时可能会出现失败。这种转换失败可能由多种原因引起,以下是一些可能的原因:
- 版本差异: ENSEMBL数据库中的ID和SYMBOL可能会随着数据库版本的更新而变化。如果使用的版本不同,可能会导致转换失败。(通常是小数点问题)
- 没有对应关系: 有些ENSEMBL ID可能没有对应的SYMBOL,尤其是在某些物种中,或者因为一些基因还没有被正式命名。(尤其是非模式生物,这些物种的数据库记录非常惨淡)
- 多对一关系: 有时候多个ENSEMBL ID 可能映射到同一个SYMBOL,这种情况下转换可能会不明确。
- 一对多关系: 有时候一个ENSEMBL ID 可能会对应多个不同的SYMBOL,尤其是在复杂基因家族中。
- 基因重命名: 基因的命名可能会发生变化,例如,之前的一个SYMBOL被废弃,然后被替换成了新的SYMBOL,这可能导致转换失败。
- 物种差异: 不同物种的基因命名和ID分配规则可能不同,可能会导致在跨物种转换时出现问题。
- 数据不完整或错误: 有时ENSEMBL数据库中的信息可能存在缺失、错误或不一致的情况,这可能导致转换失败。
- 使用的工具或数据库不准确: 如果使用的转换工具或数据库中的数据不够准确或不全面,可能会导致转换失败。
为了解决这些问题,进行ID转换时建议采取以下步骤:
- 确保使用的ENSEMBL版本和SYMBOL数据是最新的。
- 考虑使用多个转换工具或数据库进行比较,以确保结果的准确性。
- 在进行ID转换前,先检查目标数据库是否支持转换。
- 了解源数据和目标数据之间的差异,特别是在物种、版本和命名上。
- 在转换结果中,始终保留原始的ENSEMBL ID作为备用。
- 在转换过程中,随时检查和验证结果,以确保准确性。
总之,ID转换在生物信息学中是一个常见的任务,但由于多种因素的影响,转换可能会出现失败或不准确的情况。仔细考虑和验证转换结果,选择合适的工具和数据库,可以帮助降低转换失败的风险。
大家可以看到其实整个解决方案都是在chatGPT辅助下
没办法说chatGPT不重要了,当然了前提是我们有基本的生物信息学判断能力,chatGPT说好帮手,但是不能替代你哦!
如果你还没办法使用chatGPT,不妨考虑一下我们的基本的使用及密钥充值请见推文:好物安利 | 国内可稳定使用的GPT-4,还自带联网功能噢。大家对BioinfoArk使用的一些疑问也整理成了腾讯文档实时更新的形式给到大家