常规我们检测单细胞水平的肿瘤拷贝数分析方法主要是基于单细胞转录组数据,尤其是10x技术的单细胞转录组,最经典的当然是inferCNV啦,但是方法学上面也一直有更新。比如 2022的文章:《sciCNV: high-throughput paired profiling of transcriptomes and DNA copy number variations at single-cell resolution》
它提出来了 single-cell inferred chromosomal copy number variation (sciCNV), a tool for inferring single-cell CNVs from scRNA-seq at 19-46 Mb resolution.
还有《RNAseqCNV: analysis of large-scale copy number variations from RNA-seq data.》,最主要的原因并不是因为转录组数据更容易推断单细胞水平的肿瘤拷贝数,仅仅是目前市面上转录组数据是最丰富的,不得已为它开发了大量的算法。实际上转录组数据目前有两个信息可以供算法进行拷贝数的推断,比如:CaSpER uses expression values and B-allele frequencies (BAFs) from RNA-seq reads to estimate CNV events. 具体可以看2020的文章:《CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data》
拷贝数虽然说理论上连续值,但实际上大家并不会使用算法推断的连续值,反而是把它离散化,比如 5-state Hidden Markov Model (HMM) 算法:
- 1: homozygous deletion,
- 2: heterozygous deletion,
- 3: neutral,
- 4: one-copy-amplification,
- 5: high-copy-amplification.
最经典的当然是inferCNV啦,类似的算法还有:
- Single CEll Variational ANeuploidy analysis (SCEVAN)
- Clonal Architecture with Integration of SNV and CNV (CAISC), an R package for scRNA-seq data analysis
- SCYN (Single Cell and dYNamic programming), an effecient and effective dynamic programming approach for single cell data CNV segmentation and checkpoint detection.
- inferCNV
- CopyKat by Gao et al 2020
- CaSpER by Harmanci et al, 2020
- HoneyBadger by Fan et al
推断拷贝数其实是为了鉴别恶性肿瘤细胞
因为恶性肿瘤细胞的一个特性是染色体不稳定性,表现出来就是有拷贝数变化,所以我们推断拷贝数后就可以间接判断目标单细胞是否是恶性肿瘤细胞。但是特性并不是共性,也就是说有很多恶性肿瘤细胞其实也可以不发生染色体上面的拷贝数变化,那么这个时候鉴别恶性肿瘤细胞还有另外的两个方法:
- 恶性通路的激活
- 癌基因活抑癌基因的表达量失调
但是这两个方法目前并没有统一的代码和标准,所以大家看到鉴别恶性肿瘤细胞仍然是推断拷贝数的方法学,尤其是最经典的inferCNV,实际上早期的inferCNV方法学在各个癌症单细胞转录组数据应用的时候都是跟配对的肿瘤外显子数据做对比,来说说明inferCNV的准确性,也就是说,肿瘤外显子这样的DNA层面的测序数据才是更权威的。不过肿瘤外显子其实重要的分析还是在看somatic突变信息里面的SNV,这些分析我们五年前就都系统性梳理过:
- 使用R包deconstructSigs根据已知的signature进行比例推断
- 使用R包SomaticSignatures进行denovo的signature推断
- 下载TCGA所有癌症的maf文件做signature分析
- TCGA各大癌症的somatic突变全景图鸟瞰(动态交互)
- 根据肿瘤突变信息计算一系列指标
- 你真的理解体细胞突变吗?
- 突变位点生存分析
- 比较不同的肿瘤somatic突变的signature
- 一步一步用Snakemake搭建gatk4生成正常样本的germline突变数据库的流程
- 学徒作业-两个基因突变联合看生存效应
- 多位点取样外显子测序看食管癌的肿瘤内部突变异质性(逆向收费读文献2020-15)
- SNV突变(96种)频谱的制作
DNA层面的CNV更可靠
虽然早期我们仅仅是做单细胞转录组层面数据,但是目前已经是有单细胞DNA测序(scDNA-seq)技术啦,最初的基于全基因组扩增 (WGA) 的单细胞测序方法,一次仅限于分析几个到几十个细胞。随着基于微滴、纳米孔和组合索引方法的发展,极大地提高了细胞通量并降低了成本。这个领域MD安德森癌症研究中心的Nicholas Navin课题组一直走在前面:
- 空间单细胞DNA测序技术:Topographic Single Cell Sequencing 发表于2018年CELL杂志,文章题目是:Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing31449-6)
- 2021年Nature文章中报道的适用于新鲜或冰冻样本的单细胞DNA扩增方法ACT
- 文章Archival single-cell genomics reveals persistent subclones during DCIS progression,报道了第一个能对FFPE组织进行高通量单细胞DNA测序的方法:Arc-well (archive nanowell sequencing),发表在 2023年8月15日的 CELL杂志
论文中作者首先评估了Arc-well在新鲜样本(未福尔马林固定)的技术表现。通过与其他5个scDNA-seq技术:ACT, 10X CNV, DLP, DLP+ 和DOP-PCR相比,Arc-well技术显示了最低的数据分布偏好性,且在相同数据量的情况下获得了最高的基因组覆盖度。
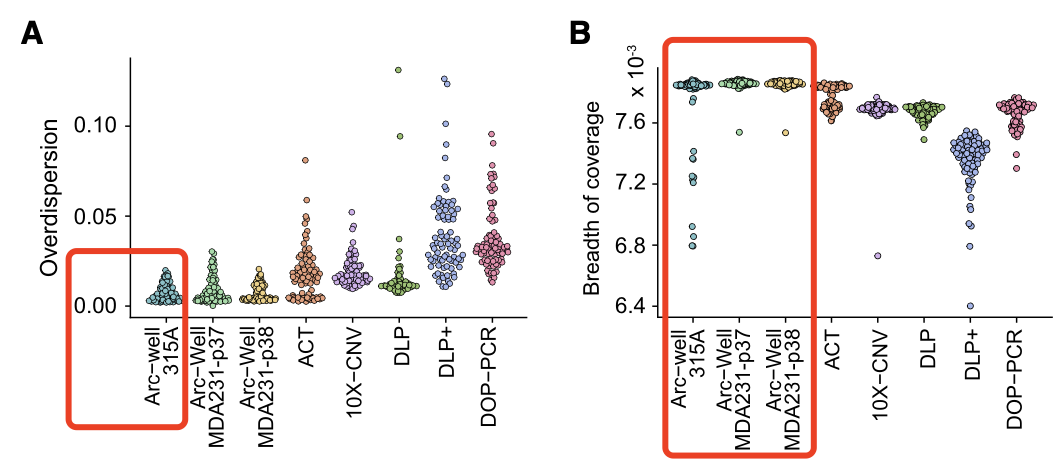
作者对存放了3-31年(平均约20年)的来自于22个乳腺癌、2个肺癌和2个前列腺癌的的FFPE样本进行了Arc-well测序并发现Arc-well获得了不错的数据质量,且都能有效的检测各个组织中存在的单细胞水平的拷贝数变异。虽然尔马林固定的样品导致了数据质量的稍微下降,但在检测拷贝数变异方面并无任何差别。
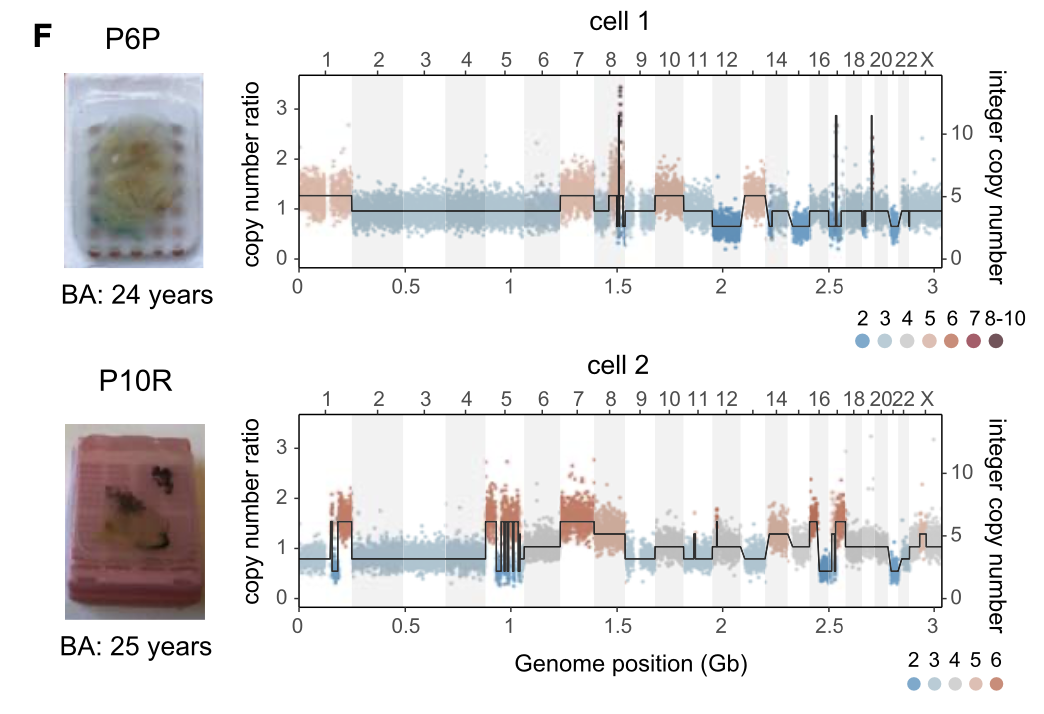
通过比较原发和复发克隆的祖先细胞中存在的拷贝数变异,作者鉴定了每一对样本中复发样本中特异存在的拷贝数事件。通过整合分析,作者还鉴定了存在于复发样本中的高频拷贝数变异事件,比如chr3q (PIK3CA) 、chr5p 、 chr8p (MYC, CCNE2) 、 chr9q 和chr20q (ZNF217, AURKA) 的扩增等,而这些拷贝数变异很可能导致了癌症的复发。