前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。
其中 GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,科学家通常是使用来超几何分布检验这个统计学算法做富集分析,即通过比较实际观察到的基因集合(几十个或者几百个)中特定功能或通路的基因数量与随机期望的数量来判断其是否富集。
接下来,让我们演示一下如何针对一个转录组数据进行差异分析后挑选统计学显著的上下调基因后进行KEGG数据库注释,最后使用pathview增强你的KEGG数据库注释结果的可视化。
首先是简单的转录组差异分析
首先我们获取airway这个包里面的airway表达量矩阵,它有8个样品,分成了两组,我们命名为’control’,’case’ ,如下所示代码:
library(data.table)
library(airway,quietly = T)
data(airway)
mat <- assay(airway)
keep_feature <- rowSums (mat > 1) > 1
ensembl_matrix <- mat[keep_feature, ]
library(AnnoProbe)
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
group_list = as.character(airway@colData$dex);group_list
group_list=ifelse(group_list=='untrt','control','case' )
group_list = factor(group_list,levels = c('control','case' ))
然后针对上面的airway这个包里面的airway表达量矩阵,是一个简单的转录组测序后的count矩阵,可以走DESeq2进行转录组差异分析,代码如下所示:
(colData <- data.frame(row.names=colnames(symbol_matrix),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = symbol_matrix,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds,
contrast=c("group_list",
levels(group_list)[2],
levels(group_list)[1]))
resOrdered <- res[order(res$padj),]
DEG =as.data.frame(resOrdered)
DEG_deseq2 = na.omit(DEG)
差异分析的结果矩阵比较简单:
> head(DEG_deseq2)
baseMean log2FoldChange lfcSE stat pvalue padj
SPARCL1 997.2841 4.601648 0.21173761 21.73278 1.005191e-104 1.751345e-100
STOM 11193.6206 1.450654 0.08458760 17.14973 6.314974e-66 5.501289e-62
PER1 776.4988 3.183045 0.20133158 15.80996 2.656240e-56 1.542656e-52
PHC2 2738.0273 1.386358 0.09161873 15.13182 9.988847e-52 4.350892e-48
MT2A 3656.0130 2.202678 0.14718400 14.96547 1.234488e-50 4.301697e-47
DUSP1 3408.8006 2.948154 0.20174509 14.61326 2.311720e-48 6.712850e-45
上面的这些列的简要介绍是:
- baseMean:
- 表达水平的均值,表示基因在不同条件或组合中的平均表达水平。
- log2FoldChange:
- 表达差异的对数倍数变化,表示基因在两个条件或组合之间的表达变化倍数的对数。
- lfcSE:
- 对数倍数变化的标准差,用于衡量差异的稳定性。
- stat:
- 统计检验的值,通常是log2FoldChange除以lfcSE,用于判断基因表达的变化是否显著。
- pvalue:
- 统计检验的p值,表示在零假设下观察到的差异或效应发生的概率。
- padj:
- 经过多重检验校正后的p值,用于控制多重检验的误差,提高差异的可靠性。
然后使用最流行的clusterProfiler进行KEGG数据库的注释
前面的DESeq2进行转录组差异分析后的表格里面有两万多个基因,但是我们根据里面的log2FoldChange对基因排序后取 log2FoldChange 最大的1000个基因来使用超几何分布检验这个统计学算法做富集分析,代码如下所示:
DEG_deseq2=DEG_deseq2[order(DEG_deseq2$log2FoldChange),]
up_top_genes = AnnotationDbi::select(org.Hs.eg.db,keys=tail(rownames(DEG_deseq2),1000) ,
columns="ENTREZID",keytype = "SYMBOL")[,2]
library(clusterProfiler)
library(org.Hs.eg.db)
kk.up <- enrichKEGG(gene = up_top_genes,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.up)[,1:6]
dotplot(kk.up)
kk.up=DOSE::setReadable(kk.up, OrgDb='org.Hs.eg.db',
keyType='ENTREZID')#按需替换
head(kk.up@result)
可以看到这个结果表格非常的丰富,但是我们通常是给一个平平无奇的条形图,可以看到最显著的一些通路的名字以及其对应的富集信息:
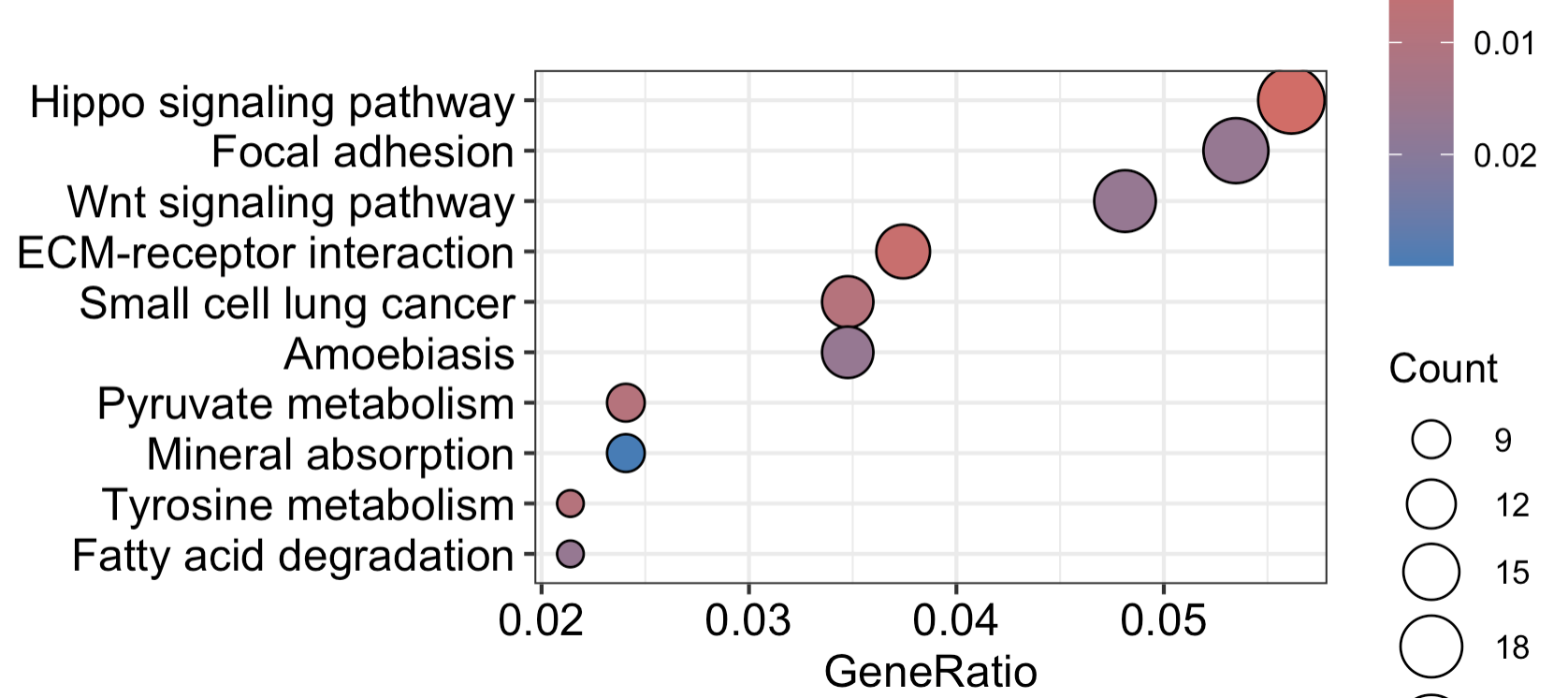
如果我们有感兴趣的通路,其实可以使用pathview增强你的KEGG数据库注释结果的可视化,比如上面的 Hippo signaling pathway 通路,
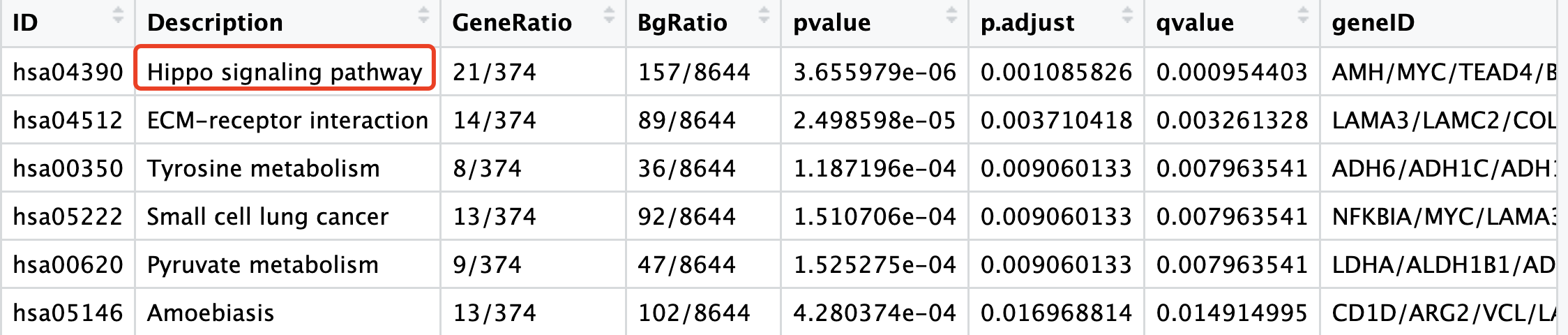
如果是我们想看看这个通路里面的全部的基因具体是哪些表达量上调哪些下调,就可以使用如下所示的代码:
#BiocManager::install('pathview')
library(pathview)
# hsa04390 Hippo signaling pathway
fc = DEG_deseq2$log2FoldChange
names(fc) = rownames(DEG_deseq2)
head(fc)
p <- pathview(gene.data = fc ,
pathway.id = "04390", species = "hsa",
out.suffix = "airway", kegg.native = T, gene.idtype = "SYMBOL")
str(p)
可以很明显的看到,这个 Hippo signaling pathway 通路里面的基因确实是绝大部分都是上调的:
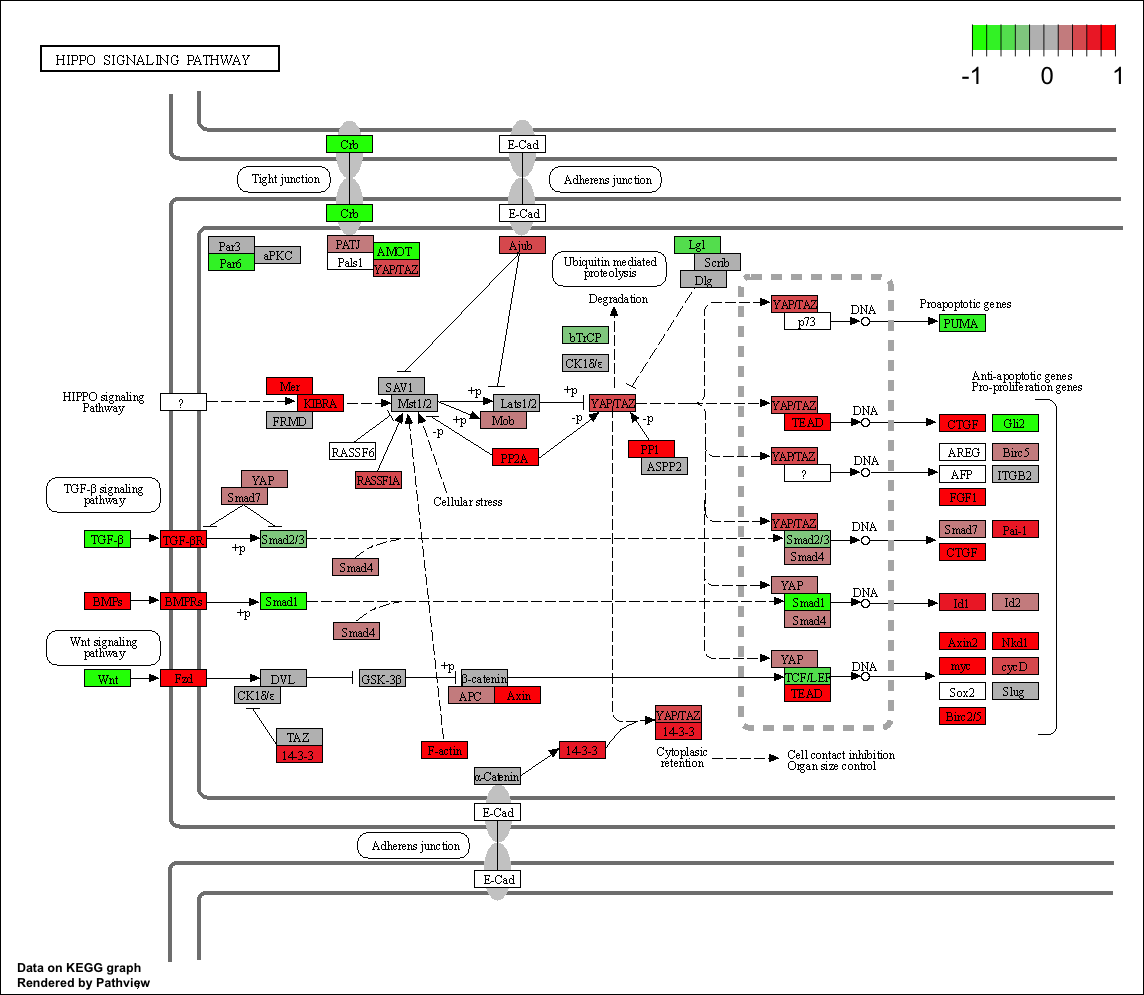
值得注意的是上面的代码里面有一个参数是kegg.native = T,说的是我们出图的时候保留kegg官网的布局,其实也可以试试看让它为F,会得到如下所示的pdf格式的图片哦!
