前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。
其中 GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,但是GO数据库 注释通常包括三个方面的信息:分子功能(Molecular Function)、细胞组分(Cellular Component)和生物过程(Biological Process)。而前面我们演示了:使用topGO增强你的GO数据库注释结果的可视化,是超几何分布检验的结果的可视化,主要是展示GO数据库的有向无环图结构。接下来我们聊聊使用clusterProfiler的GSEA方法针对GO数据库进行注释后的结果的可视化,所以是需要大家自己提前弄清楚GSEA方法和超几何分布检验方法的区别哦!
首先是简单的转录组差异分析
首先我们获取airway这个包里面的airway表达量矩阵,它有8个样品,分成了两组,我们命名为’control’,’case’ ,如下所示代码:
library(data.table)
library(airway,quietly = T)
data(airway)
mat <- assay(airway)
keep_feature <- rowSums (mat > 1) > 1
ensembl_matrix <- mat[keep_feature, ]
library(AnnoProbe)
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
group_list = as.character(airway@colData$dex);group_list
group_list=ifelse(group_list=='untrt','control','case' )
group_list = factor(group_list,levels = c('control','case' ))
然后针对上面的airway这个包里面的airway表达量矩阵,是一个简单的转录组测序后的count矩阵,可以走DESeq2进行转录组差异分析,代码如下所示:
(colData <- data.frame(row.names=colnames(symbol_matrix),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = symbol_matrix,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds,
contrast=c("group_list",
levels(group_list)[2],
levels(group_list)[1]))
resOrdered <- res[order(res$padj),]
DEG =as.data.frame(resOrdered)
DEG_deseq2 = na.omit(DEG)
差异分析的结果矩阵比较简单:
> head(DEG_deseq2)
baseMean log2FoldChange lfcSE stat pvalue padj
SPARCL1 997.2841 4.601648 0.21173761 21.73278 1.005191e-104 1.751345e-100
STOM 11193.6206 1.450654 0.08458760 17.14973 6.314974e-66 5.501289e-62
PER1 776.4988 3.183045 0.20133158 15.80996 2.656240e-56 1.542656e-52
PHC2 2738.0273 1.386358 0.09161873 15.13182 9.988847e-52 4.350892e-48
MT2A 3656.0130 2.202678 0.14718400 14.96547 1.234488e-50 4.301697e-47
DUSP1 3408.8006 2.948154 0.20174509 14.61326 2.311720e-48 6.712850e-45
上面的这些列的简要介绍在前面演示的有:使用topGO增强你的GO数据库注释结果的可视化
然后使用clusterProfiler的GSEA方法针对GO数据库进行注释
前面的DESeq2进行转录组差异分析后的表格里面有两万多个基因,需要对它们根据里面的log2FoldChange对基因排序后的全部的基因的列表,而前面我们演示了:使用topGO增强你的GO数据库注释结果的可视化,是超几何分布检验的结果的可视化只需要统计学显著的上下调的几百个基因即可。
代码如下所示:
nrDEG=DEG_deseq2[,c("log2FoldChange", "padj")]
colnames(nrDEG)=c('logFC','P.Value')
library(org.Hs.eg.db)
library(clusterProfiler)
gene <- bitr(rownames(nrDEG), fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
nrDEG = nrDEG[rownames(nrDEG) %in% gene$SYMBOL,]
nrDEG$ENTREZID = gene$ENTREZID[match(rownames(nrDEG) , gene$SYMBOL)]
# https://www.ncbi.nlm.nih.gov/gene/?term=SPARCL1
geneList=nrDEG$logFC
names(geneList)=nrDEG$ENTREZID
geneList=sort(geneList,decreasing = T)
head(geneList)
go_BP_enrich <- gseGO(geneList, OrgDb = org.Hs.eg.db, ont = 'BP')
head(go_BP_enrich@result)
产生的结果也是非常复杂,如下所示 :

默认的可视化
可以是针对具体的每个通路进行gsea可视化,如下所示代码:
enrichplot::gseaplot2( go_BP_enrich, geneSetID = go_BP_enrich@result[1,1], pvalue_table =T)
效果如下所示:
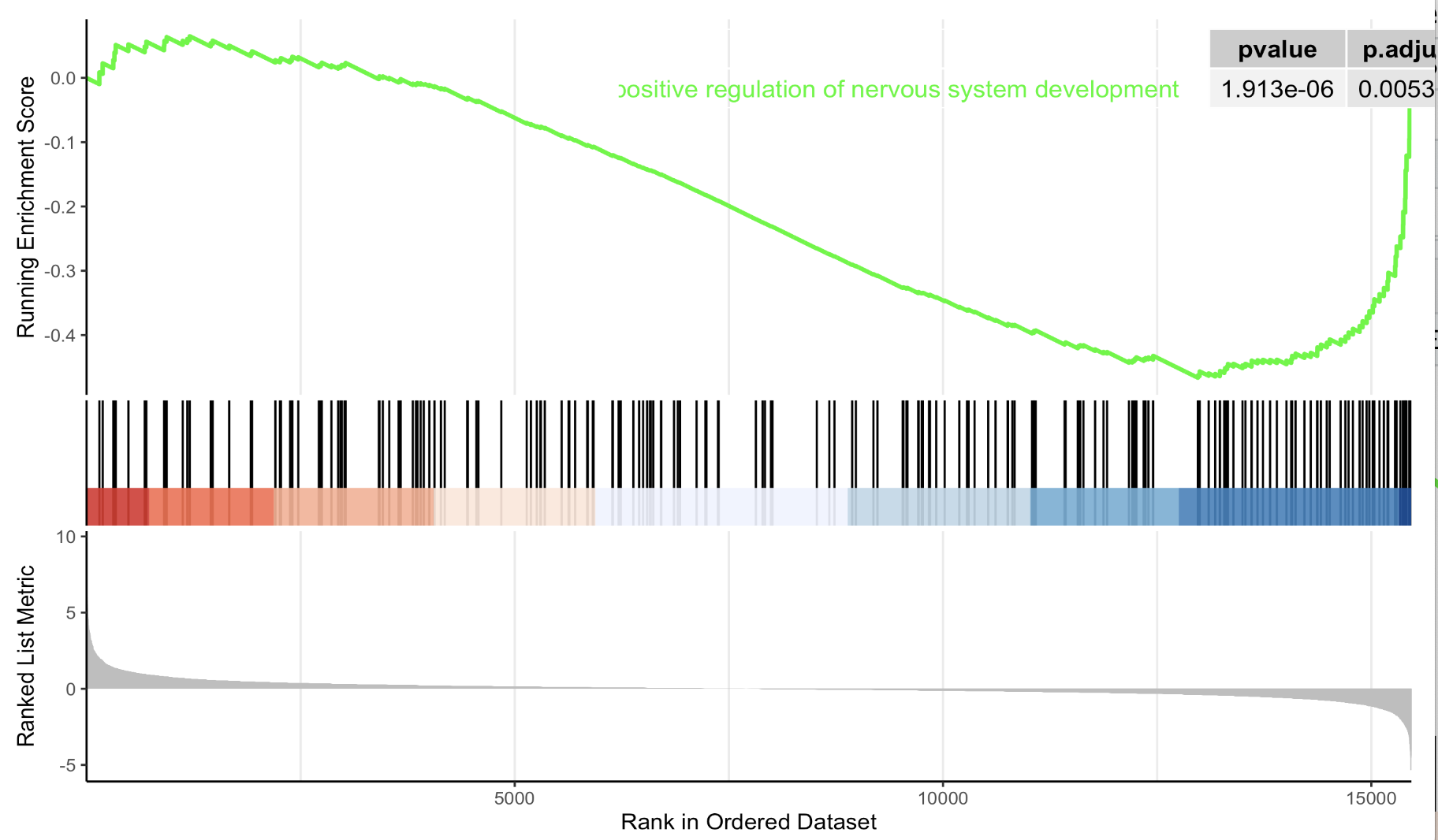
也可以是汇总这个GO数据库进行注释结果表格后展现其上下调的最显著的通路,代码如下所示:
#~~~取前6个上调通路和6个下调通路~~~
kk=go_BP_enrich
up_k <- kk[head(order(kk$enrichmentScore,decreasing = T)),];up_k$group=1
down_k <- kk[tail(order(kk$enrichmentScore,decreasing = T)),];down_k$group=-1
dat=rbind(up_k,down_k)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
p7 <- ggplot(dat, aes(x=reorder(Description,order(pvalue, decreasing = F)), y=pvalue, fill=group)) +
geom_bar(stat="identity") +
scale_fill_gradient(low="#34bfb5",high="#ff6633",guide = FALSE) +
scale_x_discrete(name ="Pathway names") +
scale_y_continuous(name ="log10P-value") +
coord_flip() +
theme_ggstatsplot()+
theme(plot.title = element_text(size = 15,hjust = 0.5),
axis.text = element_text(size = 12,face = 'bold'),
panel.grid = element_blank())+
ggtitle("Pathway Enrichment")
p7
如下所示:
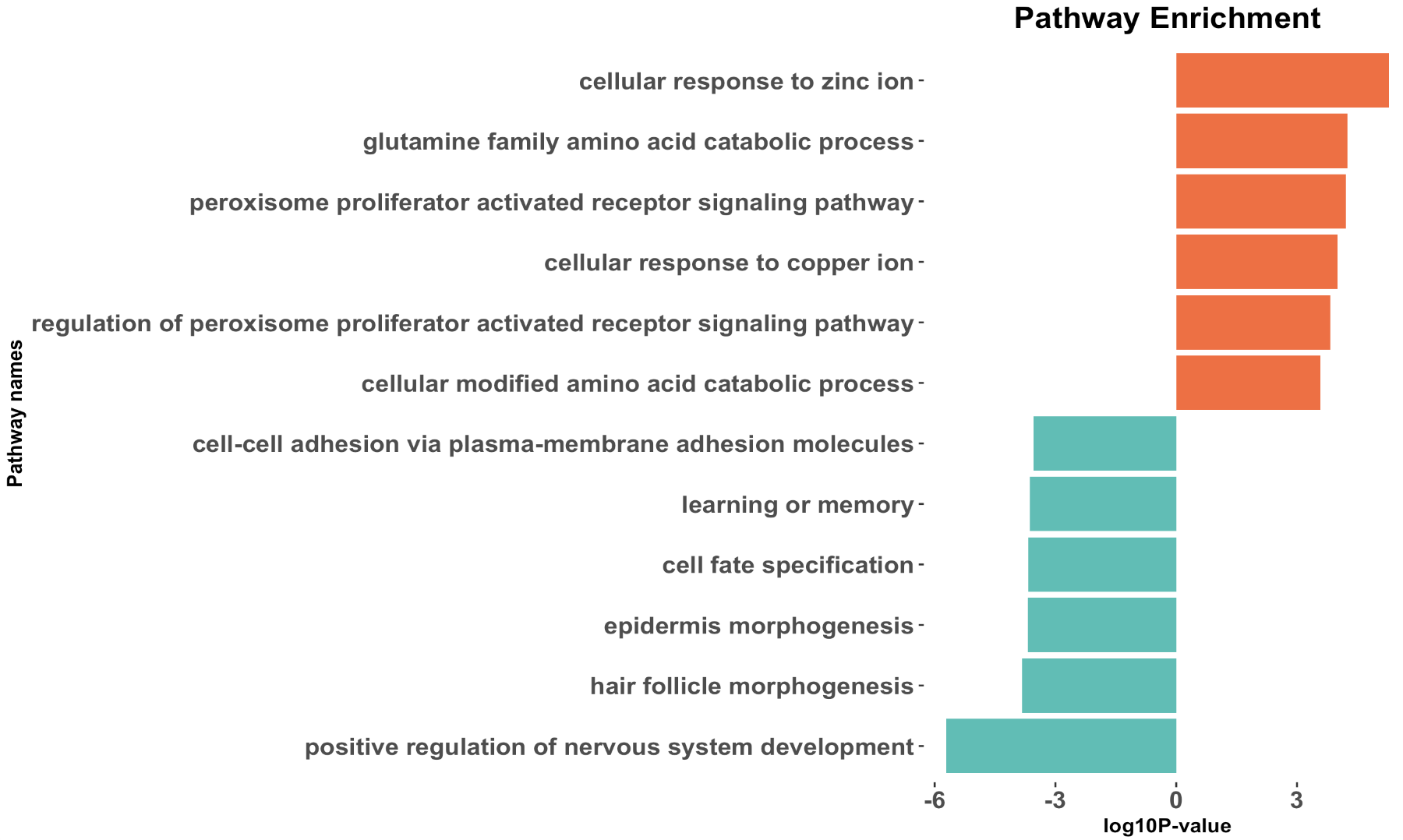
可以看到,使用clusterProfiler的GSEA方法针对GO数据库进行注释,和前面的 :使用topGO增强你的GO数据库注释结果的可视化,是超几何分布检验的结果,差异很多哦!
最后使用aPEAR来增强
同样的,也是一行代码:
#install.packages("aPEAR")
library(aPEAR)
enrichmentNetwork(go_BP_enrich@result)
可以看到,确实是效果不一样了,这也就是为什么网络可视化非常受大家欢迎。
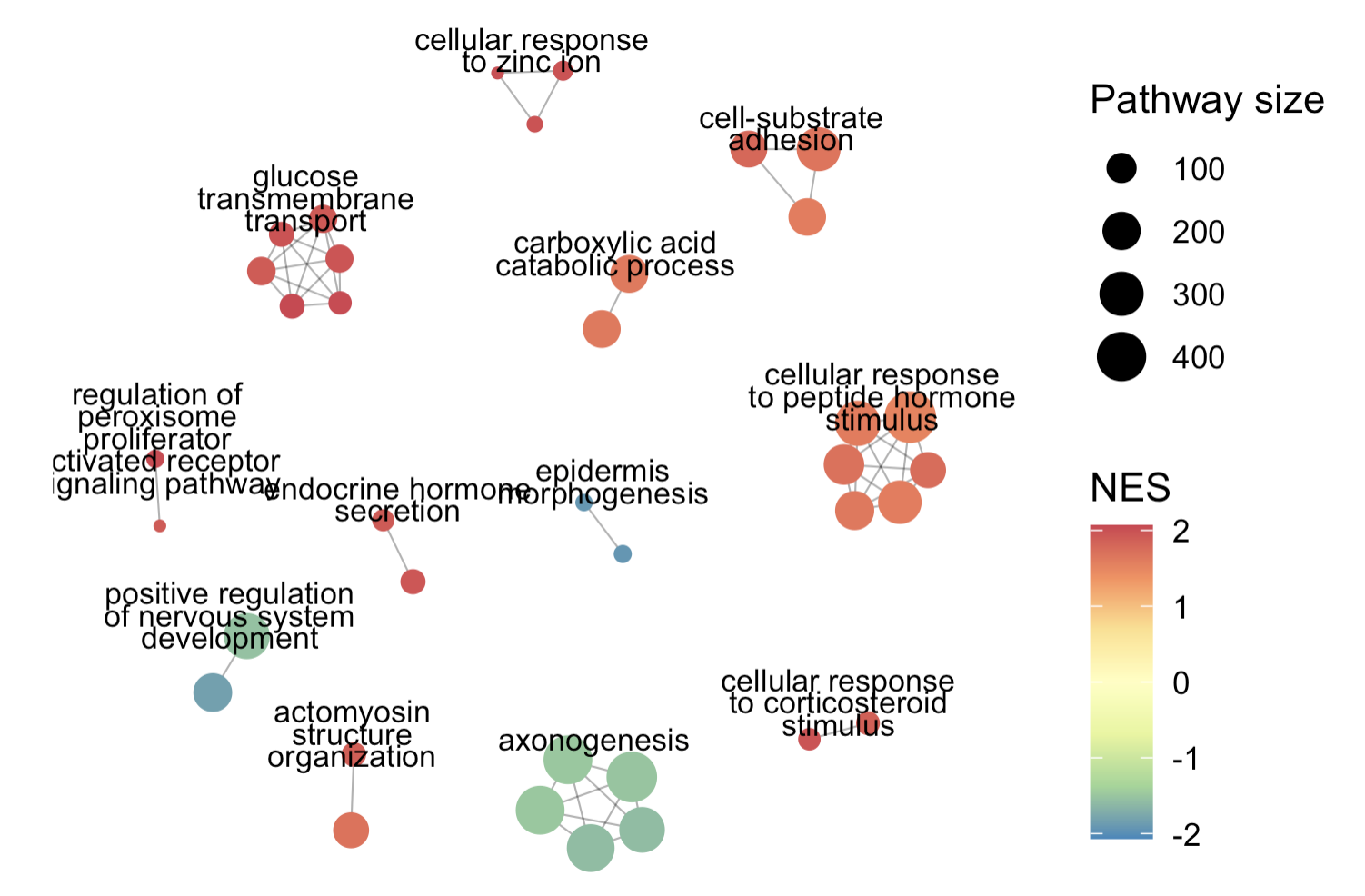
当然了,这个包(aPEAR)肯定是不会就这一个 enrichmentNetwork 函数,更多好玩的用法和参数,可以看官方文档:
- https://github.com/kerseviciute/aPEAR
- aPEAR: an R package for enrichment network visualisation
- aPEAR: an R package for autonomous visualisation of pathway enrichment networks. BioRxiv. 2023 Mar 29; doi: 10.1101/2023.03.28.534514