本笔记会被收录于《生信技能树》公众号的《单细胞2024》专辑,而且我们从2024开始的教程都是基于Seurat的V5版本啦,之前已经演示了如何读取不同格式的单细胞转录组数据文件,如下所示:
但是其它代码基本上就跟Seurat早期的v4没有区别,比如harmony整合多个单细胞样品。
但实际上Seurat有了自己的创新,官网文档:
- https://satijalab.org/seurat/articles/seurat5_integration
- https://satijalab.org/seurat/articles/integration_introduction
里面提到了它内置了多种整合多个单细胞样品的算法,可以 Perform streamlined (one-line) integrative analysis
- Anchor-based CCA integration (method=CCAIntegration)
- Anchor-based RPCA integration (method=RPCAIntegration)
- Harmony (method=HarmonyIntegration)
- FastMNN (method= FastMNNIntegration)
- scVI (method=scVIIntegration)
如果是一次性读取了多个10x样品
这个时候,因为函数Read10X可以一次性读取多个合理的路径,所以我们会把多个样品就被统一读取成为了一个稀疏矩阵而不是每个样品独立的稀疏矩阵,如下所示;
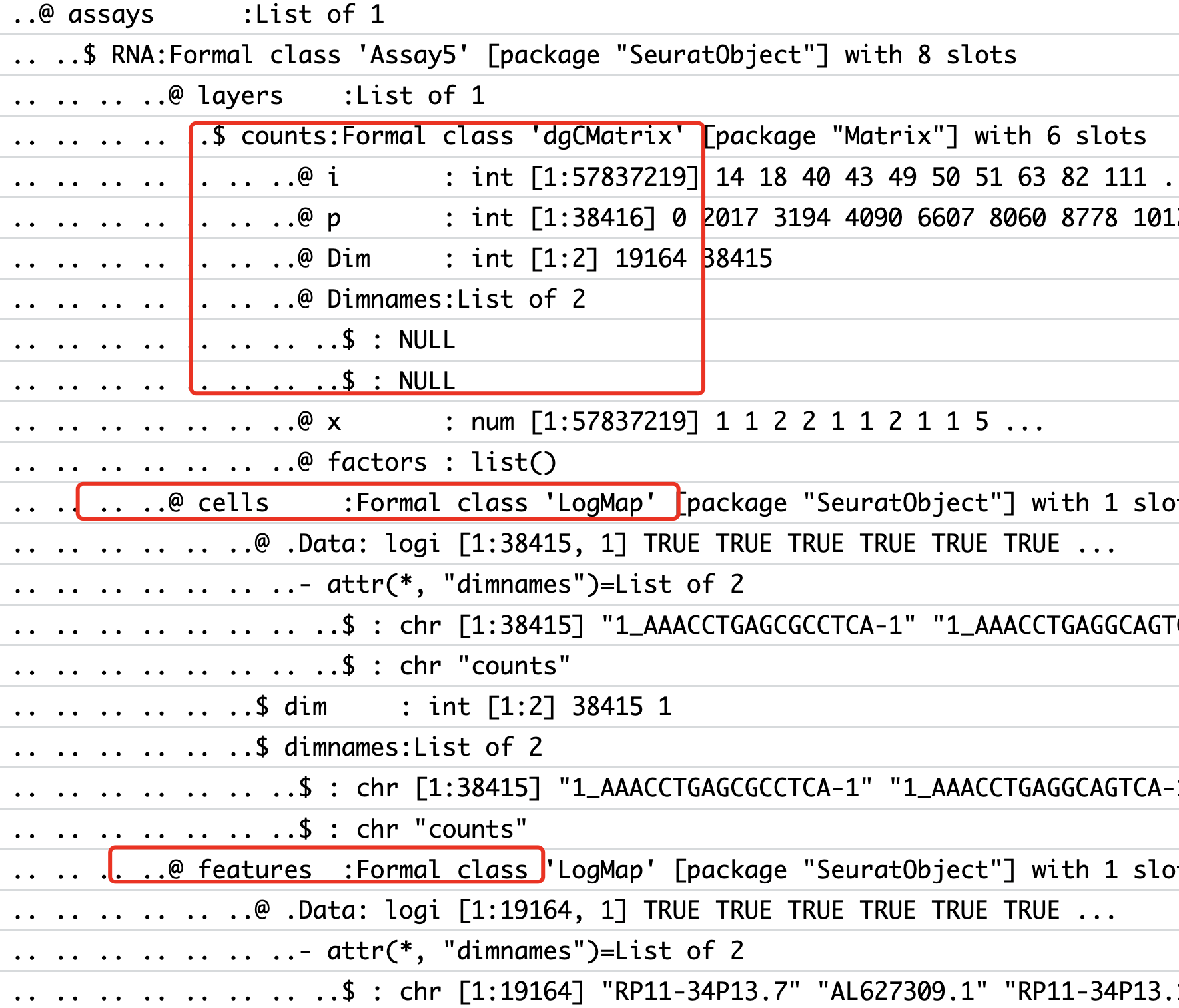
详见:使用Seurat的v5来读取多个10x的单细胞转录组矩阵,它就不适合走Seurat的v5的内置的多个单细胞样品的整合算法,所以我们会先split它,代码如下所示:
table(sce.all$orig.ident)
obj = sce.all
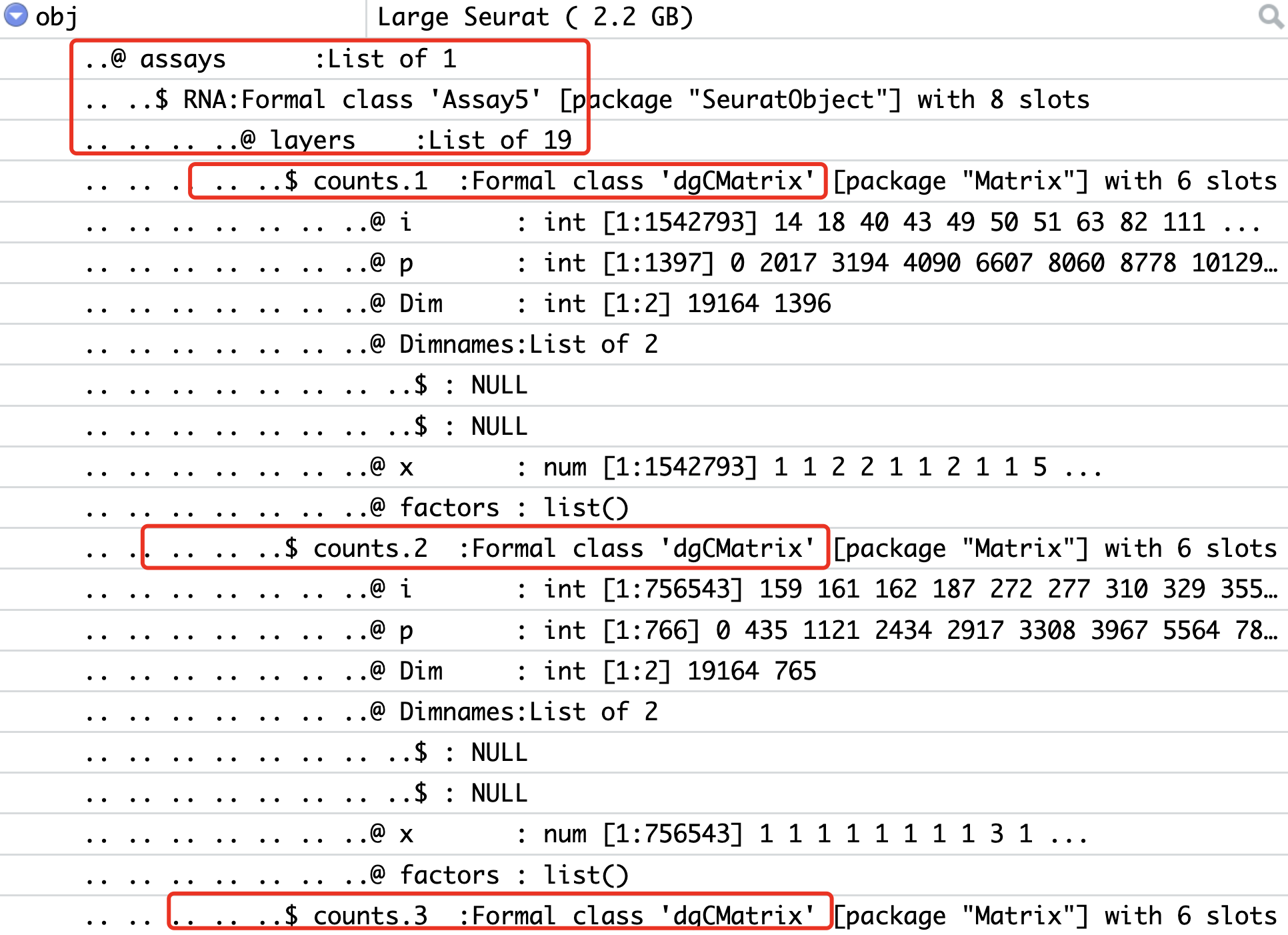
obj[["RNA"]] <- split(obj[["RNA"]], f = obj$orig.ident)
效果如下所示,可以看到每个样品的矩阵这个时候被上面的split函数拆开了:
接下来,如下所示走内置的harmony流程,就是IntegrateLayers函数里面的 :
obj <- NormalizeData(obj)
obj <- FindVariableFeatures(obj)
obj <- ScaleData(obj)
obj <- RunPCA(obj)
# 运行的harmony需要基于上面的PCA结果哦
sce.all <- IntegrateLayers(
object = obj, method = HarmonyIntegration,
orig.reduction = "pca", new.reduction = "harmony",
verbose = FALSE
)
sce.all@reductions$harmony
sce.all <- FindNeighbors(sce.all, reduction = "harmony", dims = 1:30)
res = c(0.01, 0.05, 0.1, 0.2, 0.3, 0.5,0.8 )
sce.all <- FindClusters(sce.all, resolution = res )
sce.all <- RunUMAP(sce.all, reduction = "harmony", dims = 1:30)
p1 <- DimPlot( sce.all,label = T)
p1
是很容易看到harmony被成功运行啦。
如果是先自己跑RunHarmony函数
这个时候又不能用split函数拆开了的Seurat对象哦,需要最开始那个多个样品就被统一读取成为了一个稀疏矩阵的Seurat对象,这个时候可以命名为 input_sce 变量,然后走下面的代码:
print(dim(input_sce))
input_sce <- NormalizeData(input_sce,
normalization.method = "LogNormalize",
scale.factor = 1e4)
input_sce <- FindVariableFeatures(input_sce)
input_sce <- ScaleData(input_sce)
input_sce <- RunPCA(input_sce, features = VariableFeatures(object = input_sce))
seuratObj <- RunHarmony(input_sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
# p = DimPlot(seuratObj,reduction = "umap",label=T )
# ggsave(filename='umap-by-orig.ident-after-harmony',plot = p)
input_sce=seuratObj
input_sce <- FindNeighbors(input_sce, reduction = "harmony",
dims = 1:15)
这个效果跟前面的IntegrateLayers函数是一回事,如果你这个时候的Seurat对象里面的矩阵是按照样品拆开的, 就需要先合并才能走RunHarmony函数,它来自于harmony这个r包啦。
通常是不建议走内置的harmony流程
其实我就不应该介绍这个IntegrateLayers函数的,因为它需要split那个矩阵,这样的话后面的很多分析都会有问题,比如我们跑 cosg 函数针对那个矩阵去找marker就会遇到报错:
# remotes::install_github('genecell/COSGR')
# genexcell <- Seurat::GetAssayData(object = object[[assay]],slot = slot)
marker_cosg <- cosg(
sce.all.int,
groups='all',
assay='RNA',
slot='data',
mu=1,
n_genes_user=100)
如下所示:
Error in `Seurat::GetAssayData()`:
! GetAssayData doesn't work for multiple layers in v5 assay.
Run `rlang::last_trace()` to see where the error occurred.
> rlang::last_trace()
<error/ You can run 'object <- JoinLayers(object = object, layers = layer)'.>
Error in `Seurat::GetAssayData()`:
! GetAssayData doesn't work for multiple layers in v5 assay.
---
Backtrace:
▆
1. └─COSG::cosg(...)
2. ├─Seurat::GetAssayData(object = object[[assay]], slot = slot)
3. └─SeuratObject:::GetAssayData.StdAssay(object = object[[assay]], slot = slot)
Run rlang::last_trace(drop = FALSE) to see 1 hidden frame.
>
如果要解决这个报错, 就需要首先把前面的split的矩阵重新joint回去,又是麻烦的事情!!!