PDX模型(Patient-Derived Xenograft Model)对肿瘤研究的小伙伴来说非常熟悉了,这样的癌症研究模型,它涉及将来自患者的癌细胞或组织移植到小鼠等动物宿主体内培养。这种模型的目的是更好地模拟人体内癌症的特性,以进行更真实、更有效的药物测试和研究。
在建立PDX模型时,通常是将来自患者的肿瘤组织移植到小鼠中,这种肿瘤组织中包含了患者原发癌症的细胞。因此,取样时主要涉及的是人体内的肿瘤组织,但是同时也会有小鼠细胞混合,如果拿去做单细胞转录组建库测序,得到的测序数据里面就会有两个物种。
cellranger是一个常用的工具,特别适用于处理10x Genomics平台生成的数据。如果你的样品来源于人和鼠的混合细胞,你需要考虑到物种差异,以便在进行分析时正确识别和区分来源于人和鼠的细胞。首先需要安装了cellranger软件,你可以从10x Genomics官方网站上下载并安装最新版本。同样的,在官方网站可以下载到含人和鼠的混合基因组的参考文件,来自Illumina测序的fastq文件准备好,并确保它们按照cellranger的要求进行命名。接下来就可以运行cellranger count命令即可拿到表达量矩阵文件。
解析表达量矩阵文件
矩阵文件需要按照每个样品独立的文件夹,并且文件夹里面是3个文件,如下所示:
tree -h ../inputs/
[ 160] ../inputs/
├── [ 160] s1
│ ├── [ 34K] barcodes.tsv.gz
│ ├── [670K] features.tsv.gz
│ └── [ 71M] matrix.mtx.gz
└── [ 160] s2
├── [ 66K] barcodes.tsv.gz
├── [670K] features.tsv.gz
└── [124M] matrix.mtx.gz
就可以使用下面的默认代码进行读取啦:
dir='../inputs/'
samples=list.files( dir )
samples
# samples = head(samples,10)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce =CreateSeuratObject(counts = Read10X(file.path(dir,pro )) ,
project = gsub('^GSM[0-9]*_','',
gsub('filtered_feature_bc_matrix','',pro) ) ,# pro, #
min.cells = 5,
min.features = 500 )
print(dim(sce))
print(sce@assays$RNA@counts[1:4,1:4])
return(sce)
})
names(sceList)
sceList[[1]]
as.data.frame(sceList[[1]]@assays$RNA@counts[1:10, 1:2])
as.data.frame(tail(sceList[[1]]@assays$RNA@counts[ , 1:2]))
# GRCh38-
# mm10---
可以很清晰的看到其中一个样品如下所示:
> sceList[[1]]
An object of class Seurat
38379 features across 6379 samples within 1 assay
Active assay: RNA (38379 features, 0 variable features)
> as.data.frame(sceList[[1]]@assays$RNA@counts[1:10, 1:2])
AAACCCAAGAGTACCG-1 AAACCCAAGGTATAGT-1
GRCh38-AL627309.1 0 0
GRCh38-AL627309.5 0 0
GRCh38-AP006222.2 0 0
GRCh38-LINC01409 0 0
GRCh38-FAM87B 0 0
GRCh38-LINC01128 0 0
GRCh38-LINC00115 0 0
GRCh38-FAM41C 0 0
GRCh38-AL645608.6 0 0
GRCh38-AL645608.2 0 0
> as.data.frame(tail(sceList[[1]]@assays$RNA@counts[ , 1:2]))
AAACCCAAGAGTACCG-1 AAACCCAAGGTATAGT-1
mm10---CAAA01118383.1 0 0
mm10---Csprs 0 0
mm10---Vamp7 0 0
mm10---Tmlhe 0 0
mm10---CAAA01147332.1 0 0
mm10---AC149090.1 0 0
虽然说是6379多个细胞,有38379个基因,但是里面肯定是有人和小鼠,我们可以简单的拆分,如下所示:
sceList = lapply(sceList,function(sce){
#sce = sceList[[1]]
ct = sce@assays$RNA@counts
kp=grepl('mm10---',rownames(ct))
table(kp)
ct = ct[!kp,]
rownames(ct) = gsub('GRCh38-','',rownames(ct))
ct[1:4,1:4]
sce = CreateSeuratObject(
counts = ct, min.cells = 5,
min.features = 500
)
print(dim(sce))
print(sce@assays$RNA@counts[1:4,1:4])
return(sce)
})
# gsub('^GSM[0-9]*','',samples)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = gsub('^GSM[0-9]*_','',
gsub('filtered_feature_bc_matrix','',samples)) )
# gsub('_gene_cell_exprs_table.txt.gz','',
# gsub('^GSM[0-9]*_','',samples) )
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
# GRCh38-
# mm10---
主要是根据表达量矩阵里面的 GRCh38和mm10前缀,它们在基因名字前面,是可以筛选和切除的。上面演示的是保留人类基因名字的矩阵,简单的修改过滤的逻辑就是保留小鼠基因的表达量矩阵进行后续的降维聚类分群啦。
也可以是物种+病毒
前面的PDX模型(Patient-Derived Xenograft Model)是来源于多个物种的单细胞转录组表达量矩阵的典型例子, 其实类似的案例还有很多,比如各种癌症都有对应的病毒,如果你想探索就需要单细胞转录组定量的时候需要修改参考基因组。
而且非洲猪瘟也是如此,恰好看到了2022的文章《Transcriptome profiling in swine macrophages infected with African swine fever virus at single-cell resolution》,描述了很清楚这个定量过程:

就是前面提到的首先需要安装了cellranger软件,你可以从10x Genomics官方网站上下载并安装最新版本。然后制作两个物种的混合基因组的参考文件,来自Illumina测序的fastq文件准备好,并确保它们按照cellranger的要求进行命名。接下来就可以运行cellranger count命令即可拿到表达量矩阵文件。
它的运行cellranger count命令即可拿到表达量矩阵文件就是在数据集:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168113
GSM5129158 UninfectedGroup_T0
GSM5129159 UninfectedGroup_T2
GSM5129160 UninfectedGroup_T6
GSM5129161 UninfectedGroup_T10
GSM5129162 UninfectedGroup_T15
GSM5129163 UninfectedGroup_T24
GSM5129164 UninfectedGroup_T36
GSM5129165 InfectedGroup_T2
GSM5129166 InfectedGroup_T6
GSM5129167 InfectedGroup_T10
GSM5129168 InfectedGroup_T15
GSM5129169 InfectedGroup_T24
GSM5129170 InfectedGroup_T36
需要同样的整理它提供的表达量矩阵,然后读取后,可以看到表达量矩阵里面也是被加入了前缀 ;
> head(sce.all@assays$RNA@counts[ , 1:2])
6 x 2 sparse Matrix of class "dgCMatrix"
InfectedGroup_T10_AAACCCAAGACCTCCG-1
Georgia------MGF-360-1L 3
Georgia------MGF-360-2L 1
Georgia------KP177R .
Georgia------L83L 10
Georgia------L60L .
Georgia------MGF-360-3L .
> tail(sce.all@assays$RNA@counts[ , 1:2])
6 x 2 sparse Matrix of class "dgCMatrix"
InfectedGroup_T10_AAACCCAAGAATCTAG-1
Sus-scrofa11-ZNF594 .
Sus-scrofa11-NT5M .
Sus-scrofa11-ENSSSCG00000027662 .
Sus-scrofa11-ENSSSCG00000028362 .
Sus-scrofa11-CCDC38 .
Sus-scrofa11-ENSSSCG00000038934 .
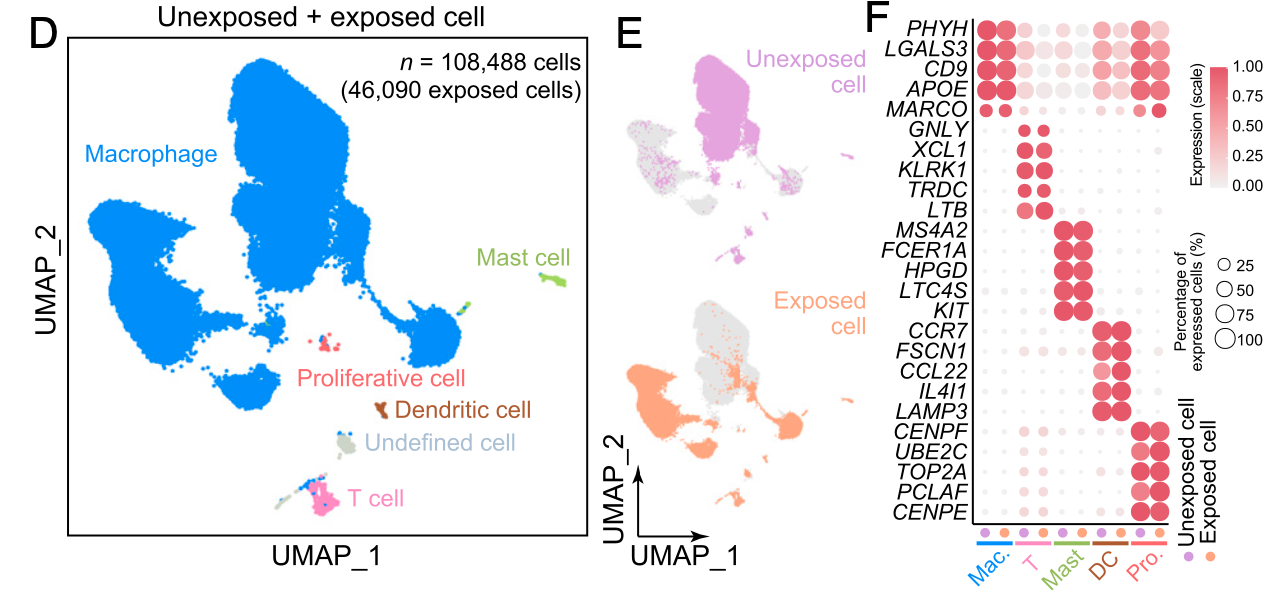
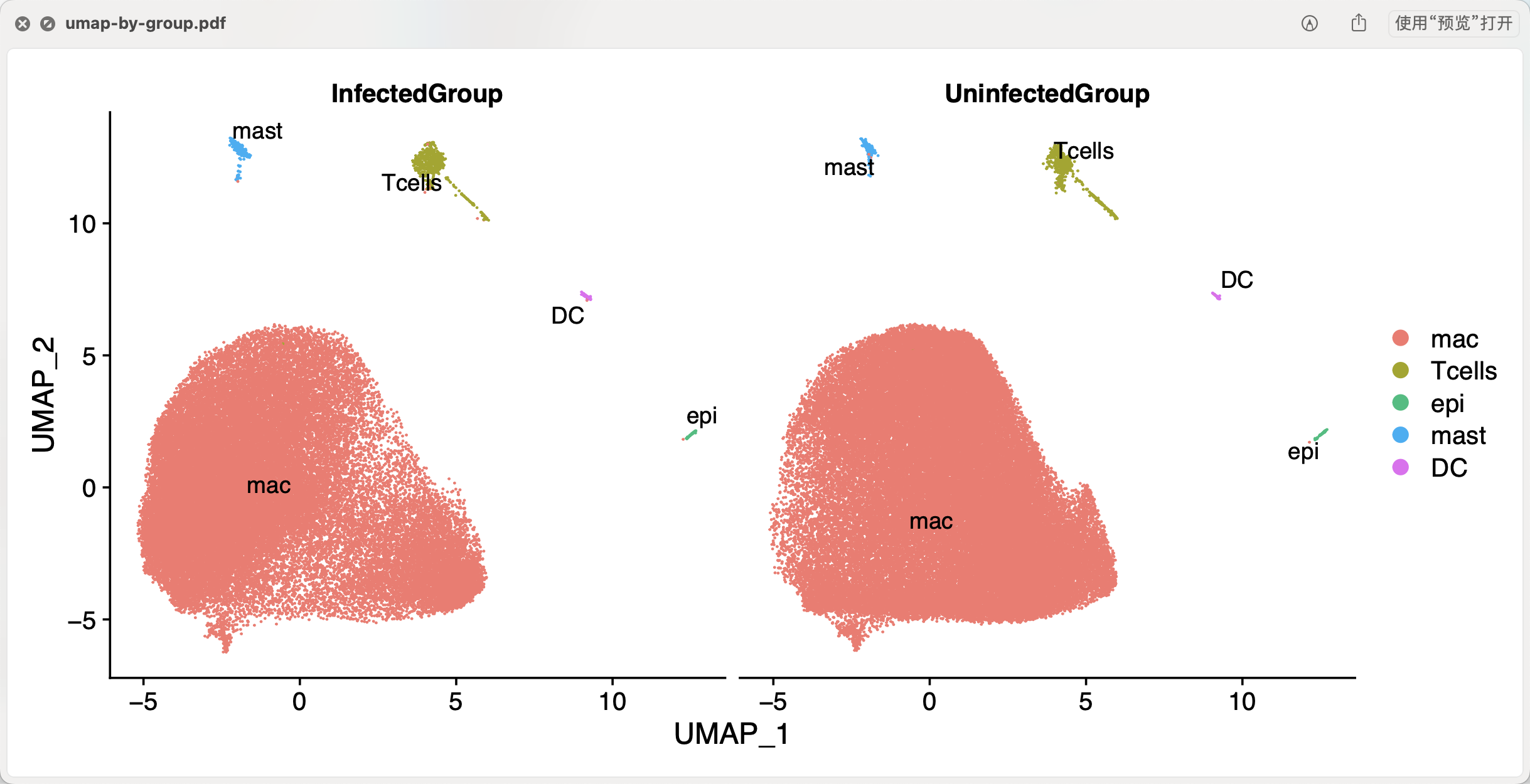
很明显就需要切割,主要的表达量矩阵进入Seurat流程进行降维聚类分群,然后很容易做的如下所示图(不过,值得注意的病毒感染与否的两个分组居然可以相差如此之大,可能是需要仔细看文章的methods描述) :
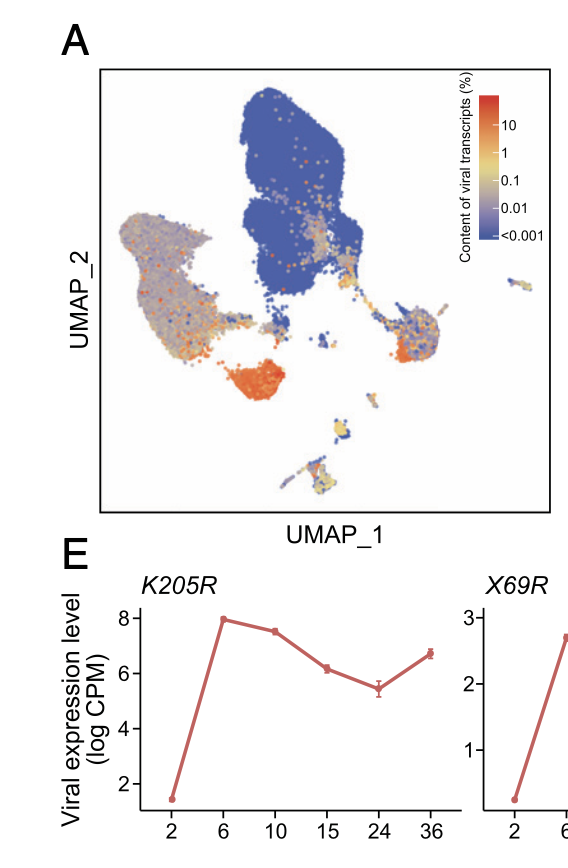
还会有一个矩阵是保存了病毒的基因表达量矩阵,就可以做丰富的叠加可视化,在前面的umap的基础上面可以把这些病毒基因表达量含量作为细胞的列属性,而不是基因表达量的行 :
如果是我们自己处理这个数据集,其实会看到每个样品的都会被很好的融合在一起,如下所示:
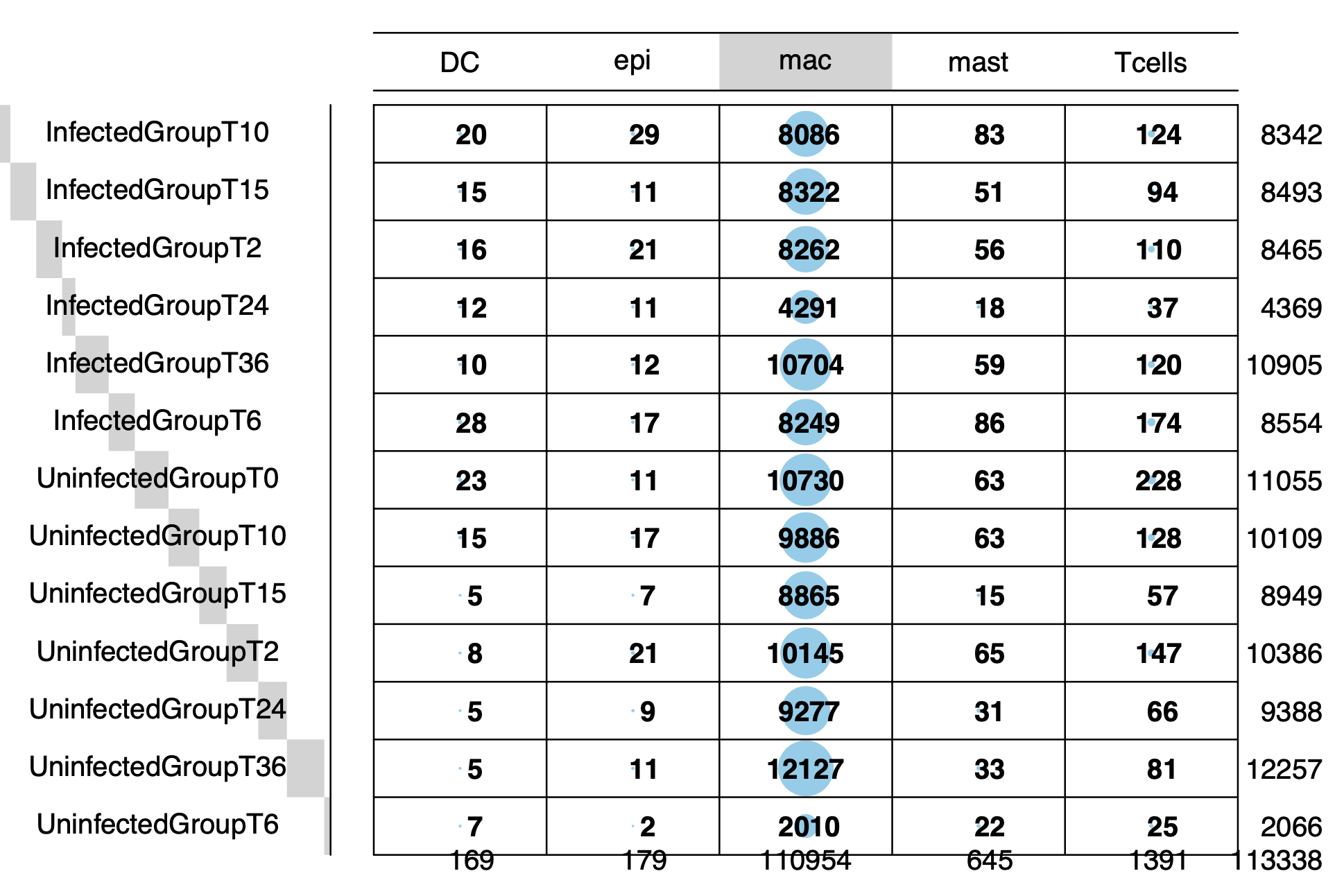
细胞数据量确实有点大:
值得思考的问题
- 为什么这个单细胞转录组数据集的降维聚类分群结果里面绝大部分细胞都是巨噬细胞呢?
- 这个数据集的物种是猪,它的巨噬细胞跟人类肿瘤里面的巨噬细胞差异很大,没有我们前面提到的M1和M2,详见:M1和M2的巨噬细胞差异就在CD86和CD163吗,而且也没有CXCL9 and SPP1的排他性,TREM2联合SPP1去和FOLR2基因的排他性。。。
- 这个数据集是两个分组各自内部多个时间点,理论上可以做pseudo-bulk 分析,也是可以根据数据分析结果拿到一个独立的生物学故事。