这个月的学徒要开始单细胞转录组结合空间单细胞的数据分析学习啦, 然后他挑选了一个文章是:《Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics》
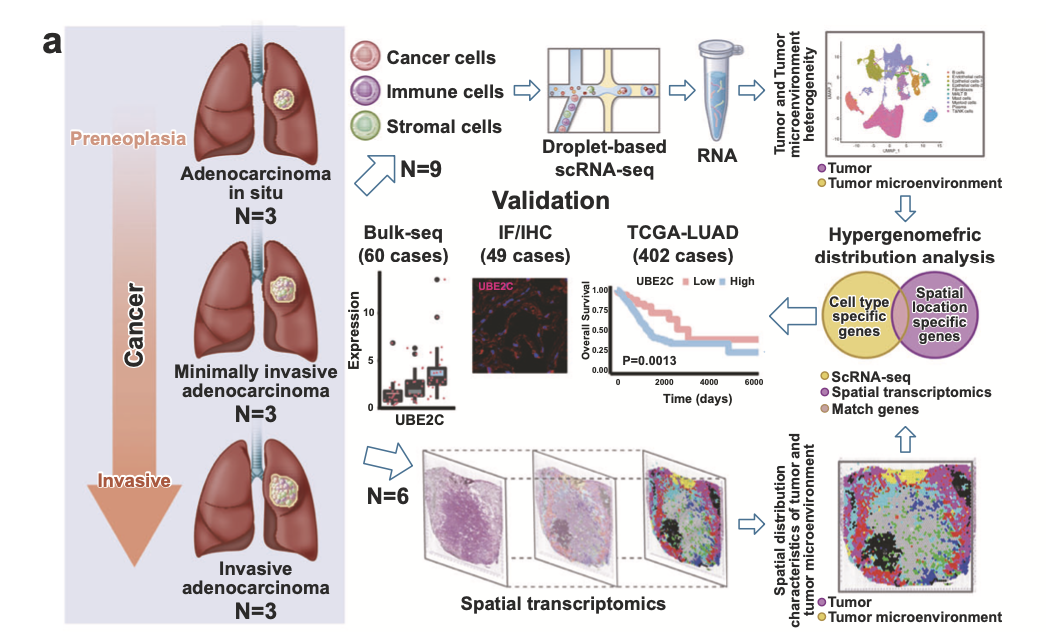
这个是2022的肺腺癌相关的文章,实验设计非常简单,就是癌症的3个发展阶段 :
- lung adenocarcinoma in situ (AIS),
- minimally invasive adenocarcinoma (MIA)
- invasive adenocarcinoma (IAC)
这3个阶段就是天然的分组啦, 每个分组是3个病人取样后做单细胞转录组,其中有两个病人的样品会同时送去做空间单细胞,所以可以看到如下所示的实验设计流程图:
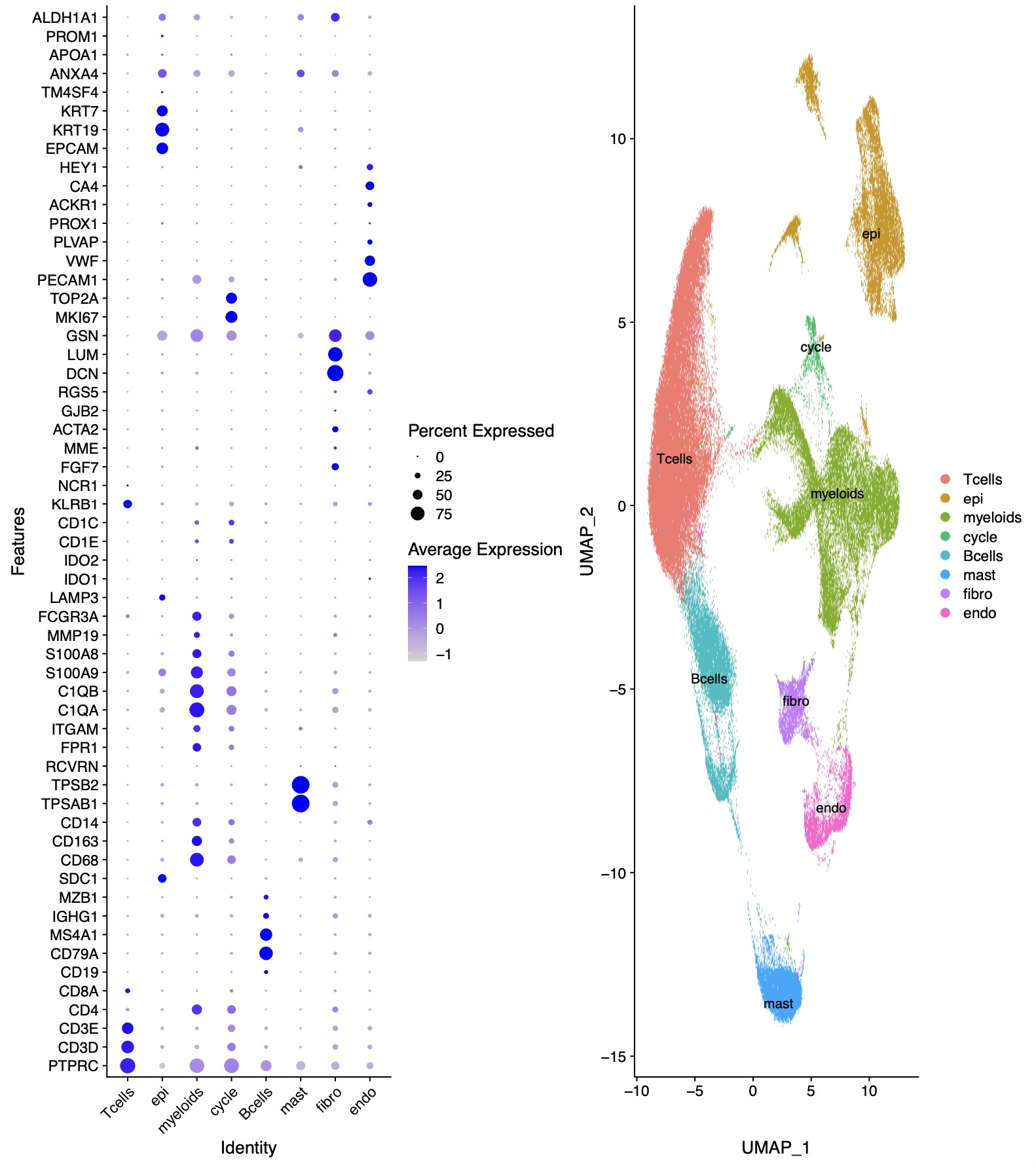
然后学徒复现文章里面的单细胞转录组数据的时候,很快就找到了数据集链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE189357,可以看到每个病人都是如下所示标准的10x单细胞转录组数据:GSM5699777_TD1_barcodes.tsv.gz 73.7 Kb GSM5699777_TD1_features.tsv.gz 297.6 Kb GSM5699777_TD1_matrix.mtx.gz 71.4 Mb肺癌单细胞数据集也有好几十个了,拿到表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。所以是很容易降维聚类分群啦,如下所示:
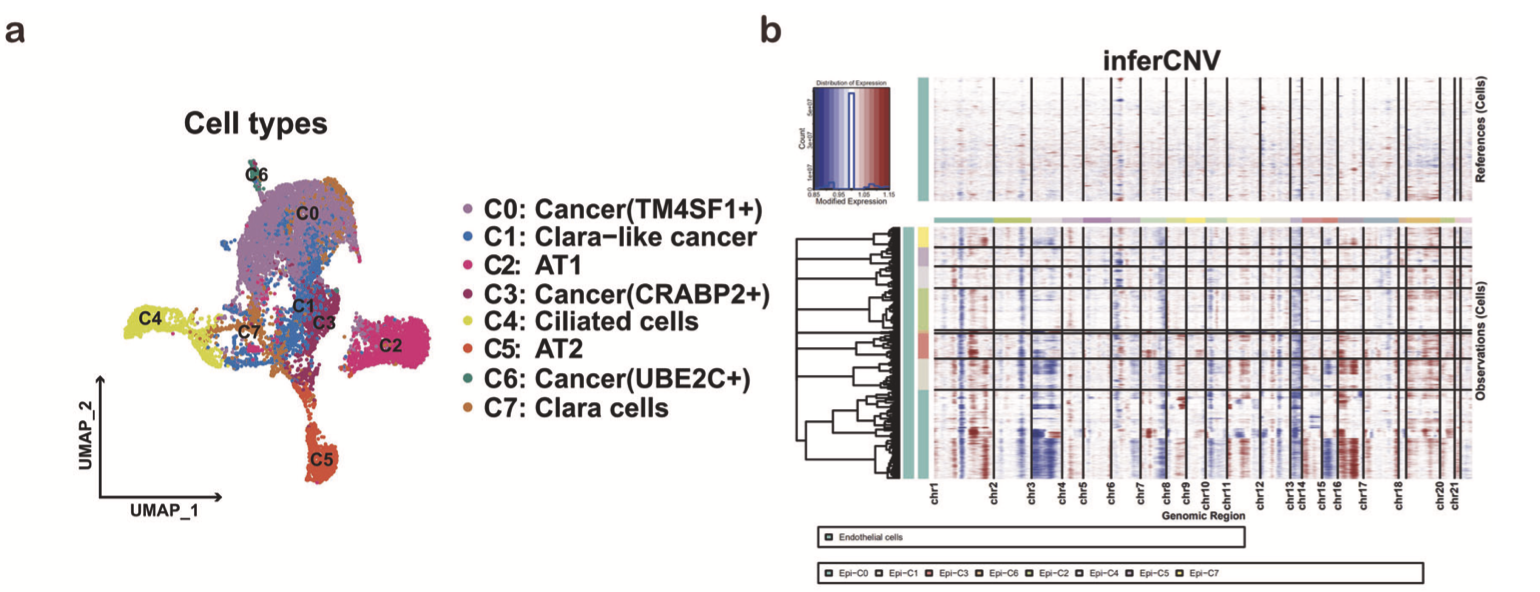
文章这个时候针对里面的上皮细胞亚群进行了细致的摸索,比如同样的降维聚类分群,并且结合拷贝数看上皮细胞亚群的细分亚群的恶性程度来判断其中有4个亚群 :
肺癌既然是来源于肺这样的组织, 它的上皮细胞就不可能是一个纯粹的上皮,理论上是可以细分的。一般来说可以分成如下所示的5个亚群: - alveolar type I cell (AT1; AGER+)
- alveolar type II cell (AT2; SFTPA1)
- secretory club cell (Club; SCGB1A1+)
- basal airway epithelial cells (Basal; KRT17+)
-
ciliated airway epithelial cells (Ciliated; TPPP3+)
如果是我们提前第一层次降维聚类分群里面的上皮细胞进行细分的时候,会发现里面仍然是混合了其它免疫细胞,如下所示可以看得到是其中 2,4,9,12,14,15 都是免疫细胞,这个就很迷惑。。。。
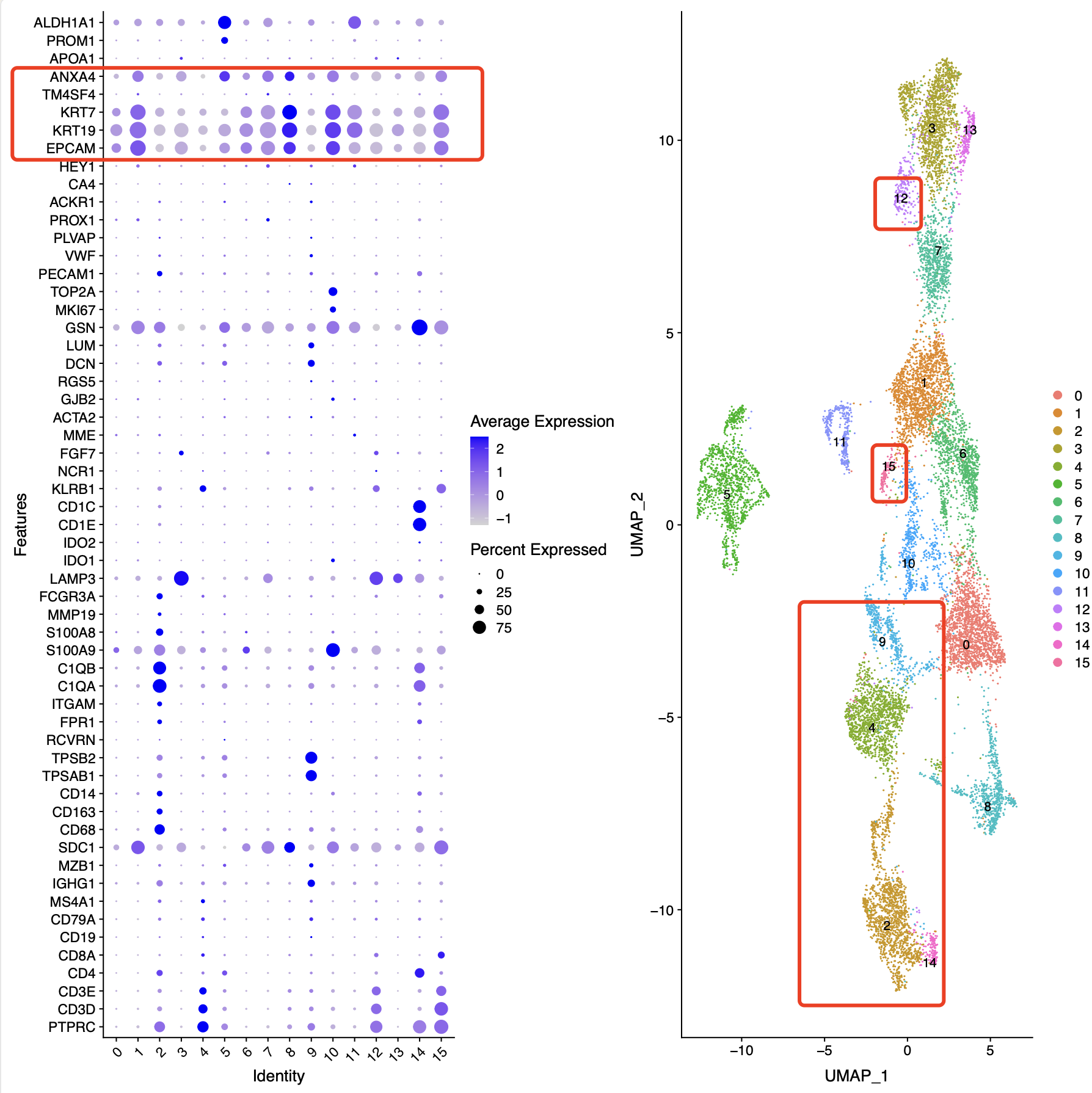
我的处理策略往往是先在上皮细胞亚群细分时候删除这些,我上面的图片显示的RNA_snn_res.0.5分辨率情况,而且因为12距离3和13太近了,我就没有删除它,这个时候很多情况下都是生信工程师自己的感觉在做数据分析。sce.all.int=sce.all.int[,!sce.all.int$RNA_snn_res.0.5 %in% c(2,4,9,14,15)] table(sce.all.int$RNA_snn_res.0.5 )然后0,1,6这3个亚群,我看不到里面的特异性高表达量基因,所以我不想给它名字 ,
celltype[celltype$ClusterID %in% c( 5 ),2]='ciliated' celltype[celltype$ClusterID %in% c(11 ),2]='secretory' celltype[celltype$ClusterID %in% c( 8 ),2]='AT2' celltype[celltype$ClusterID %in% c( 10),2]='cycle' celltype[celltype$ClusterID %in% c( 3,12,13 ),2]='LAMP3-epi' celltype[celltype$ClusterID %in% c( 0),2]='0' celltype[celltype$ClusterID %in% c( 1),2]='1' celltype[celltype$ClusterID %in% c( 6),2]='6'这样的话,跟文章里面的数据分析结果就完全不一样了,因为我可视化了文章里面的基因在我的降维聚类分群结果里面,因为我们的肿瘤细胞细分的亚群都不一样,那么就不可能说同样的差异分析和转录因子分析了:
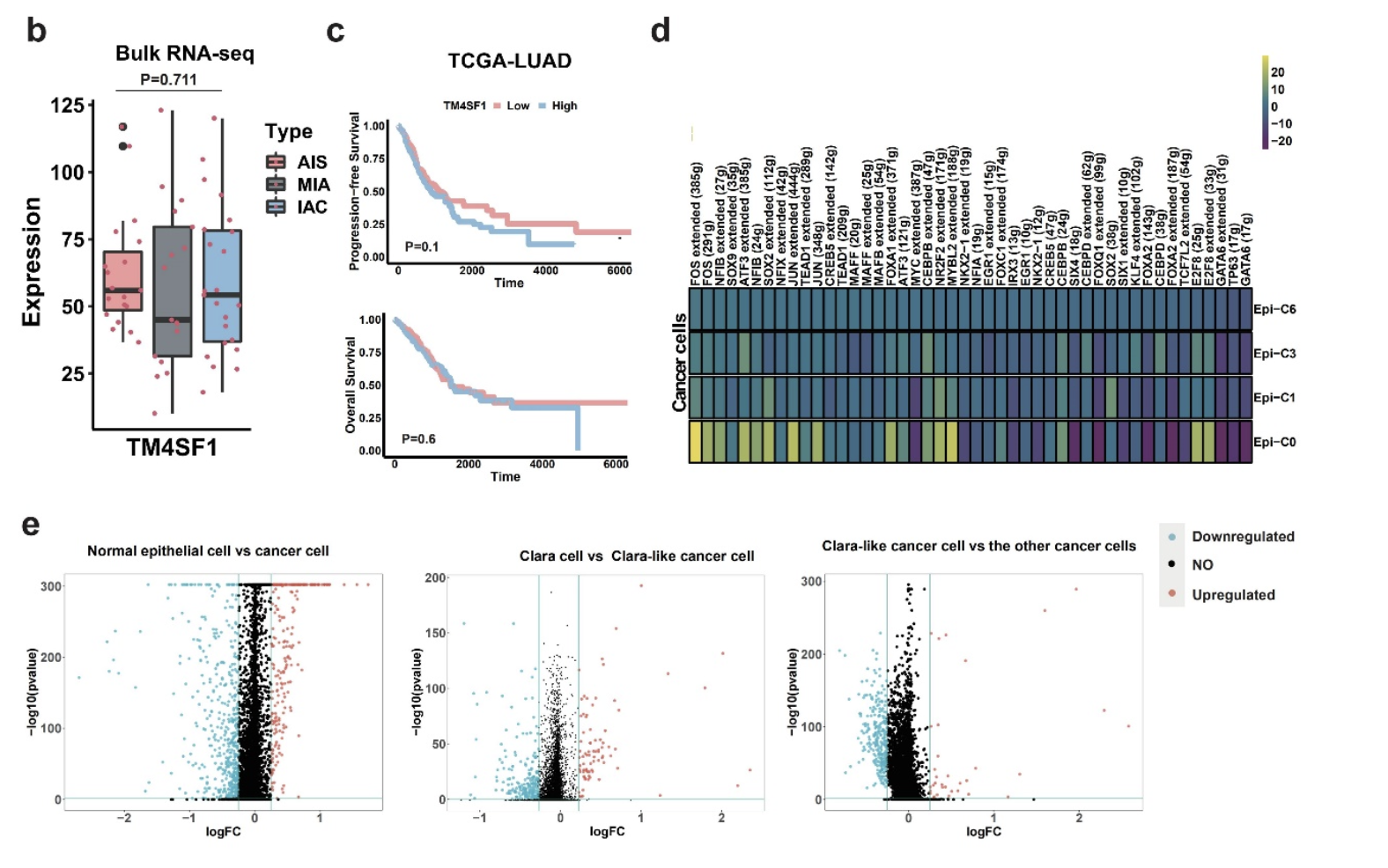
文章作者自己的多个癌细胞细分亚群都去跟正常的上皮细胞亚群进行了单细胞转录组层面的差异分分析,但是很明显其实正常上皮细胞是区分了完全不一样的功能,为什么要混合起来呢?
The other normal epithelia cells (Epi-C2, Epi-C4, Epi-C5) vs the other cancer cells (Epi-C0, Epi-C3, Epi-C6); Clara cells (Epi-C7) vs Clara-like cancer cells (Epi-C1), Clara-like cancer cells (Epi-C1) vs the other cancer cells (Epi-C0, Epi-C3, Epi-C6)
我觉得我区分的亚群的各自的恶性上皮细胞亚群的基因更有意义!
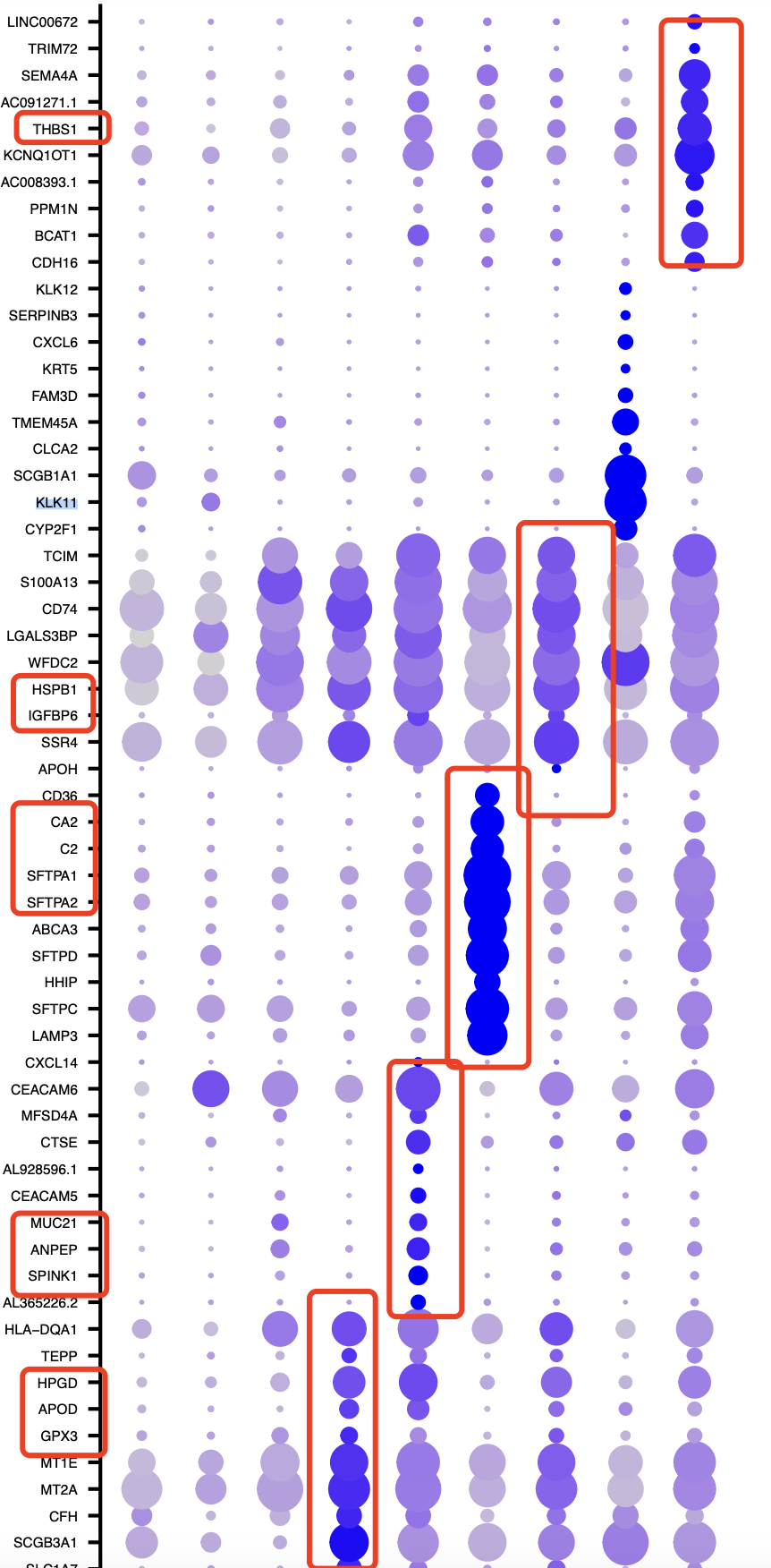
如果有认识这些基因的小伙伴,可以找我来合作进行后续分析啊!!!
因为我没有按照文章的分析策略,然后全部的结果都不一样了!所以换一个分析策略会导致文章的全部论点都得推倒重来吗?
其实并不是这样的,因为这个文章的落脚点是 UBE2C + cancer cells (Epi-C6)] 就是大名鼎鼎的肿瘤恶性增值状态的细胞亚群,这个东西除非你数据分析错误,否则它一定会出现,我们的上面的数据分析里面我就把它命名为了cycle,但是这不都是同一个东西吗,尽管是大家的数据分析流程不一样。
值得注意的是,这个文章在做三分组的9个病人的单细胞转录组的同时,也有对应的空间单细胞,但是从数据分析的结果来看,无非就是把空间单细胞转录组数据当做是了染色的切片,看了看已知的单细胞亚群的染色的片子的分布而已。。。

当然了,也有可能是我对空间单细胞的认识还不够,恰好这周四(2023年10月26日-27日)有一个进阶课程 | 「SBC&生信技能树」第十期-单细胞及空间转录组测序生信分析培训班开课啦(线上线下同步学习),感兴趣的小伙伴可以跟我一起去学习哈。申请了一下,可以在临开课前报名,详见:报名链接 (可以私聊我,我的微信在生信共享办公室出租可以找到 )学徒作业
绝大部分癌症相关单细胞文章在常规的降维聚类分群后,都会针对每个细胞亚群的基因去看它在bulk转录组表达量矩阵看看是否有变化,以及它是否能区分生存!所以我们的作业很简单, 就是完成上面的GSE189487的降维聚类分群后提取上皮细胞进行降维聚类分群,然后对每个亚群的top特异性高表达量基因集,去做差异分析和生存分析!