最近在实习生在处理一个单细胞转录组数据集的时候,发现该研究的全部10x样品里面居然有5个是无法读取的, 所以像我求助。 很有意思。文章是:《Single-cell transcriptomic analysis of the tumor ecosystems underlying initiation and progression of papillary thyroid carcinoma》:

其公开的数据链接是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184362
可以看到是 23个样品:
- 7 primary tumors,
- 6 para-tumors,
- 8 metastatic LNs
- 2 subcutaneous metastatic loci
详细信息如下所示 :
GSM5585102 Patient 1 tumor
GSM5585103 Patient 1 paratumor
GSM5585104 Patient 2 tumor
GSM5585105 Patient 2 paratumor
GSM5585106 Patient 2 left lymph node
GSM5585107 Patient 3 tumor
GSM5585108 Patient 3 paratumor
GSM5585109 Patient 3 left lymph node
GSM5585110 Patient 3 right lymph node
GSM5585111 Patient 4 subcutaneous metastase
GSM5585112 Patient 5 tumor
GSM5585113 Patient 5 paratumor
GSM5585114 Patient 5 right lymph node
GSM5585115 Patient 6 right lymph node
GSM5585116 Patient 7 right lymph node
GSM5585117 Patient 8 tumor
GSM5585118 Patient 8 paratumor
GSM5585119 Patient 9 tumor
GSM5585120 Patient 9 paratumor
GSM5585121 Patient 10 tumor
GSM5585122 Patient 10 right lymph node
GSM5585123 Patient 11 right lymph node
GSM5585124 Patient 11 subcutaneous metastase
但是下载并且整理好文件之后,发现并不是每一个样品都可以读取的!也就是说,研究者们在公开他们的数据的时候,很有可能有一点点小错误,导致了5个左右的样品是有问题,大家也可以尝试去读取看看!
这样的话,我们以前的批量读取一个文件夹下面的全部的10x的数据的代码就会报错,所以需要设置一个机制去判别它,这里我们展现tryCatch的简单实现和复杂实现方式!
简单版本
构建如下所示的一个函数:
readsce <- function (x) {
out <- tryCatch( CreateSeuratObject( Read10X(file.path('outputs/',x)) ) ,
error = function(e) NULL)
return(out)
}
# 每个10x路径,如果下面的3个文件是正常的,就可以读取
sce=readsce(x)
这个函数,就可以读取 outputs 文件夹里面的,所有的 10x单细胞数据文件夹 啦,但是必须保证每个10x路径,如果下面的3个文件是正常的,就可以读取 。
这个时候的好处是,如果这个 10x单细胞数据文件夹 里面的文件是错误的,也不会报错,不耽误这个批量操作。
本来是想尝试复杂实现方式
不知道为什么失败了:
readsce <- function(x){
sce = tryCatch(
CreateSeuratObject( Read10X(file.path('outputs/',x)) ) ,
error = function(e){
message('Caught an error!')
},
warning = function(w){
message('Caught an warning!')
},
finally = {
message('All done, quitting.')
}
)
return(sce)
}
不过,无所谓哈。
附上完整代码:
有了前面的函数读取方式,就可以批量操作这个GSE184362数据集的全部的23个样品:
- 7 primary tumors,
- 6 para-tumors,
- 8 metastatic LNs
- 2 subcutaneous metastatic loci
全部读取的代码是:
m(list = ls())
options(stringsAsFactors = F)
library(scRNAstat)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(dplyr)
dir='outputs/'
samples=list.files( dir )
samples
getwd()
readsce <- function (x) {
out <- tryCatch( CreateSeuratObject( Read10X(file.path('outputs/',x)) ) ,
error = function(e) NULL)
return(out)
}
sceList = lapply(samples,function(x){
# x=samples[3]
print(x)
sce=readsce(x)
})
kp=unlist(lapply(sceList, is.null))
sceList=sceList[!kp]
sceList
names(sceList) = samples[!kp]
读取之后肯定是需要做一些简单的解读啦!
这个时候我们仍然是借用之前的我们在《生信技能树》公众号的一个教程:这也能画?,我提到了一个很无聊的R包,名字是:scRNAstat ,它可以4行代码进行单细胞转录组的降维聚类分群,其实完全没有技术含量, 就是把 Seurat 流程的一些步骤包装成为了4个函数:
- basic_qc (查看数据质量)
- basic_filter (进行一定程度的过滤)
- basic_workflow (降维聚类分群)
- basic_markers(检查各个亚群的标记基因)
对前面的全部的读取 成功了的10x样品进行批量操作:
lapply(names(sceList) , function(x){
# x=names(sceList)[1]
print(x)
sce=sceList[[x]]
sce
dir.create( x )
sce = basic_qc(sce=sce,org='human',
dir = x)
sce
sce = basic_filter(sce)
sce = basic_workflow(sce,dir = x)
markers_figures <- basic_markers(sce,
org='human',
group='seurat_clusters',
dir = x)
p_umap = DimPlot(sce,reduction = 'umap',
group.by = 'seurat_clusters',
label.box = T, label = T,repel = T)
p=p_umap+markers_figures[[1]]
print(p)
ggsave(paste0('umap_markers_for_',x,'.pdf'),width = 12,height = 9)
})
我们随意挑选一个10x单细胞转录组数据进行查看,可以看到,这个样品基本上都是T细胞和B细胞,以及髓系免疫细胞!
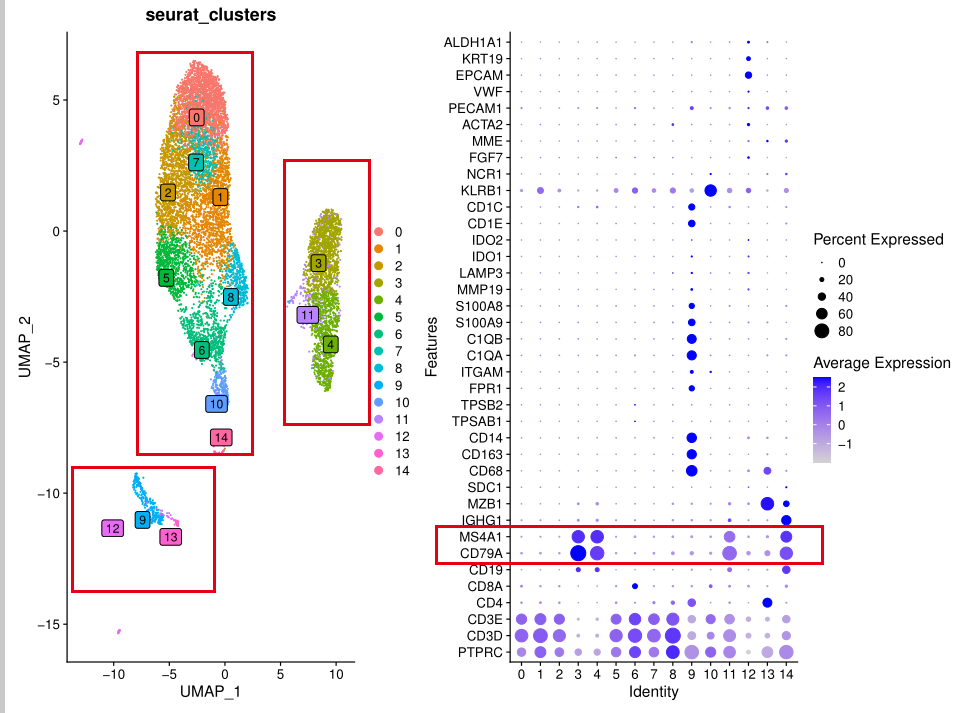
那,我们跟文章对比一下:
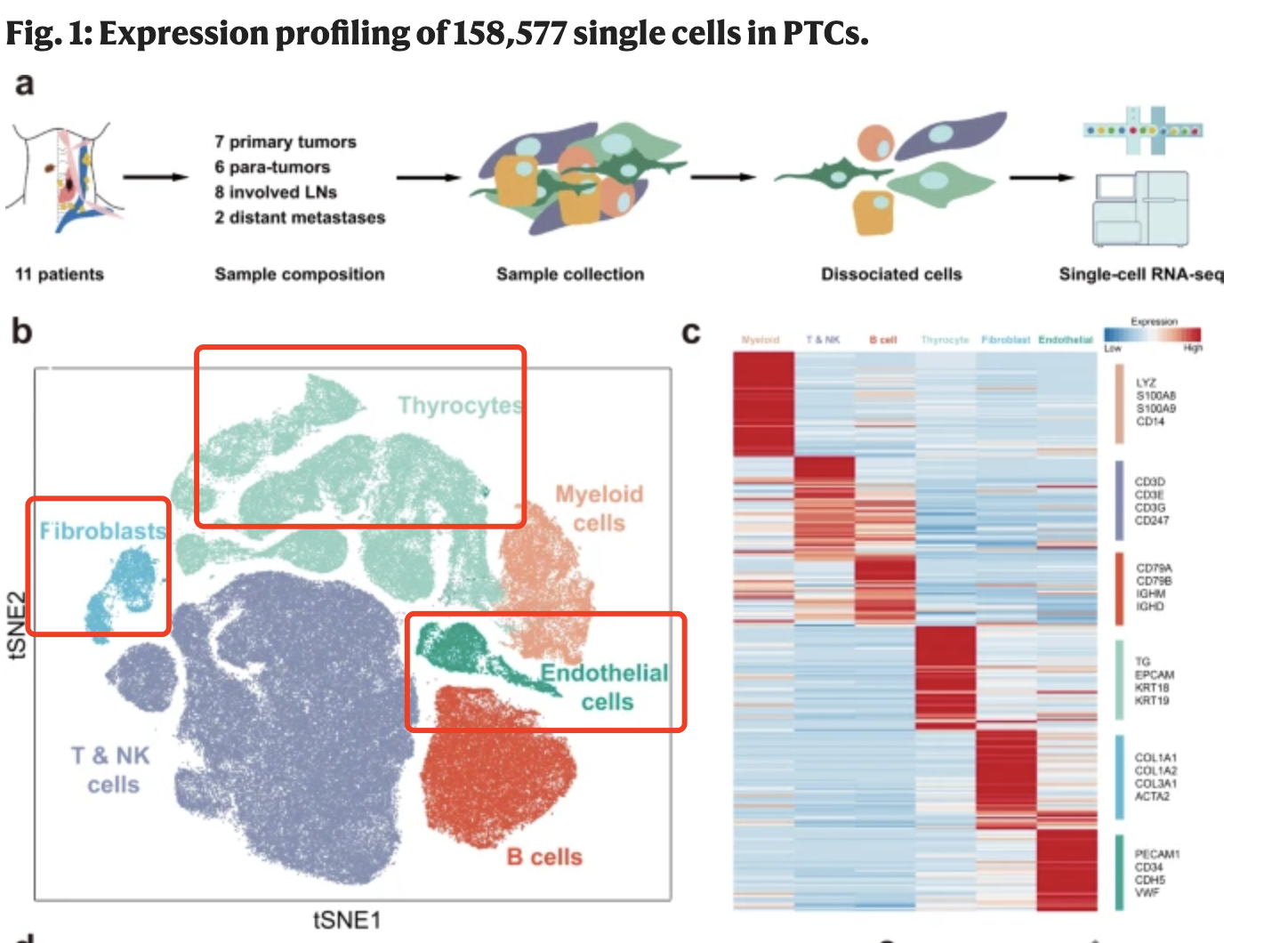
这个文章的分群基本上跟我们提到了的肿瘤单细胞分群规则类似的,首先是按照如下所示的标记基因进行第一次分群 :
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
这个文章里面比较特殊的是 thyrocytes (TG, EPCAM, KRT18, KRT19), 甲状腺细胞
如果你对单细胞数据分析还没有基础认知,可以看基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
其实单细胞测序在甲状腺癌研究中的应用文章还蛮多的,比如:
- ARHGAP36 regulates proliferation and migration in papillary thyroid carcinoma cells
- Single-cell RNA sequencing reveals a novel cell type and immunotherapeutic 2 targets in papillary thyroid cancer
- Single-cell transcriptomic landscape reveals the differences in cell differentiation and immune microenvironment of papillary thyroid carcinoma between genders
- Single-cell RNA sequencing reveals the regenerative potential of thyroid follicular epithelial cells in metastatic thyroid carcinoma